
Abstract
Gene therapy may offer a new treatment option, particularly for patients with severe hemophilia, based on recent research. However, individuals with pre-existing immunity to adeno-associated viruses (AAVs) may be less likely to benefit from AAV vector-based therapies. To study pre-existing AAV5 immunity in humans, we validated two complementary, sensitive, and scalable in vitro assays to detect AAV5 total antibodies and transduction inhibition (TI). Using these two assays, we found that 53% of samples from 100 healthy male individuals were negative in both assays, 18% were positive in both assays, 5% were positive for total antibodies but negative for TI and, of interest, 24% were negative for total antibodies but positive for TI activity, suggesting the presence of non-antibody-based neutralizing factors in human plasma. Similar findings were obtained with 24 samples from individuals with hemophilia A. On the basis of these results, we describe the development of a dual-assay strategy to identify individuals without total AAV5 antibodies or neutralizing factors who may be more likely to respond to AAV5-directed gene therapy. These assays offer a universal, transferrable platform across laboratories to assess the global prevalence of AAV5 antibodies and neutralizing factors in large patient populations to help inform clinical development strategies.
Similar content being viewed by others


Introduction
Adeno-associated virus (AAV) vector-mediated gene therapy has been investigated in human trials for the treatment of several severe diseases, including hemophilia B (HB)1, 2, 3, 4 and others.5 The results of earlier trials of AAV-mediated delivery of the human Factor IX (FIX) gene for the treatment of patients with HB demonstrated limited efficacy,3, 4 possibly because of pre-existing host humoral and cellular immunity against the AAV capsid that limits target tissue transduction and long-term expression of FIX.4, 6 Even relatively low titers of neutralizing antibodies (NAb) against the capsid have been shown to inhibit transduction of vascular-administered virus in animal models7, 8, 9, 10 and were associated with limited efficacy in human trials.4 Recent successful clinical trials of AAV8-mediated FIX gene transfer in patients with severe HB demonstrated stable, therapeutic expression of FIX in all trial participants.1, 2 Unlike previous AAV-based FIX trials, this trial excluded patients with pre-existing NAb to the AAV8 capsid as assessed in a mouse model of transduction inhibition (TI) assay using individual patient serum. These results suggest that accurate identification of subjects with pre-existing AAV immunity may be an important consideration for the design of these types of clinical trials.
Methods to detect pre-existing AAV immunity include cell-based in vitro TI assays, in vivo (for example, mice) TI assays, and enzyme-linked immunosorbent assay (ELISA)-based detection of total anti-capsid antibody (TAb) assays.6, 7 The TAb assay may be able to detect low potency NAb that are below the threshold of TI assays, but it may not detect non-antibody neutralizing factors. In vivo and in vitro TI assays screen samples for anti-AAV NAb4, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18 and other factors that modulate AAV transduction efficiency.19, 20, 21, 22, 23, 24, 25, 26 In the AAV8-FIX trial, the in vivo TI assay may have been appropriate for patient enrollment because AAV8 transduces cells in vitro poorly while it transduces mouse liver efficiently.1, 27, 28, 29 However, in contrast to the in vivo TI assay, the cell-based in vitro TI assay and TAb assay both have the advantage of being scalable, easier to standardize, and amenable to analytical validation. The utility of an in vitro cell-based TI assay was suggested in the AAV1-SERCA2a CUPID trial for patients with advanced heart failure in which the majority of subjects were negative for NAb at baseline, but subjects with detectable NAb against AAV1 may have had worse outcomes.30, 31
Another potential application for AAV-mediated gene therapy is hemophilia A (HA)—a hereditary disorder caused by a deficiency in functional clotting Factor VIII (FVIII). In order to maximize the likelihood of achieving early clinical proof-of-concept, individuals without pre-existing immunity to AAV5 could be identified using both a cell-based in vitro AAV5 TI assay and an AAV5 TAb assay. Here, we report the performance characteristics of two such assays, with details regarding statistical assay cut points (including screening, titer, and specificity cut points), specificity, selectivity, sensitivity, matrix interference, and precision. Last, we provide evidence suggesting the presence of non-antibody-based neutralizing factors to AAV5 in human plasma. This dual-assay screening strategy could be applied to AAV5-based gene therapy trials, seroprevalence studies, and trials using other AAV serotypes.
Results
TAb and TI assays used to select nonhuman primates for gene transfer
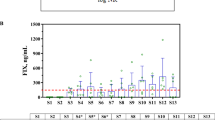
To maximize the likelihood of successful liver transduction with systemic AAV5-mediated gene transfer in nonhuman primate (NHP) pharmacology studies, we selected animals without pre-existing immunity against the AAV5 capsids. Prior to gene transfer, individual NHP plasma samples were assessed in both a cell-based TI assay and an ELISA-based AAV5 TAb assay to identify neutralizing factors and pre-existing AAV5 antibodies, respectively. The 64 NHP subjects were classified as positive or negative. Plasma that had an ELISA signal two-fold above the background in the TAb assay was designated as positive for pre-existing AAV5 antibodies. Plasma that decreased transduction of a green fluorescent protein (GFP)–AAV5 reporter virus to less than 60% of negative control plasma was designated positive in the TI assay (Figure 1a). The majority of the samples (84%) showed agreement in both assays as either double-positive (upper left quadrant) or double-negative (lower right quadrant). Twenty NHP subjects that screened negative in both assays were used in three separate AAV5-based gene transfer studies that used 3 different lots of vectors. While there was inter-subject variability observed, all individuals exhibited successful liver transduction 8 weeks post dosing, as demonstrated by vector genome (vg) detection using real-time quantitative polymerase chain reaction analysis (Figure 1b). This finding suggests that a similar approach could be beneficial for identifying human patients for clinical trials.

Prescreening of nonhuman primates (NHPs) resulted in successful liver transduction. (a) Screening of NHPs for pre-existing adeno-associated virus 5 (AAV5) immunity with NHP total anti-capsid antibody (TAb) and transduction inhibition (TI) assays. Sixty-four monkey samples were tested in both NHP AAV5 TAb and NHP AAV5 cell-based TI assays. The axes intersect at the respective assay cut points of 2.0 signal to background ratio and 60% transduction for the NHP TAb and TI assays, respectively. The majority of samples (76.6%) screened negative in both assays (lower right quadrant, n=49). Six samples (9.4%) screened positive in both assays (top left quadrant). Six samples (9.4%) screened negative in the TI assay but positive in the TAb assay (top right quadrant). Three samples (4.7%) screened positive in the TI assay but negative in the TAb assay (bottom left quadrant). (b) Detection of vector genomes in monkey liver. Twenty nonhuman primate subjects that screened negative in both assays were dosed with 2 × 1012 to 6 × 1013 vector genome (vg) per kg in three separate adeno-associated virus (AAV)-based gene transfer studies. All individuals exhibited successful liver transduction 8 weeks post dosing, as demonstrated by vector genome detection using real-time quantitative polymerase chain reaction analysis. The data are represented as vg per μg of genomic DNA (gDNA).
Assessment of assay formats to detect AAV5 TAb and TI activities in human plasma
Because of the successful implementation of a mouse in vivo AAV8 TI patient screening approach in a clinical trial for HB gene transfer,1, 27 we assessed the feasibility of a similar in vivo TI screening assay for AAV5 vector for patients with HA. During the assay development phase, mice were first injected with positive or negative control plasma (that is, NHP plasma with or without AAV5 NAb) and then with AAV5 vector carrying the FVIII transgene. The TI was assessed by measuring liver FVIII gene expression. We found that the in vivo AAV5 TI assay produced variable results and lower sensitivity than a cell-based assay (Supplementary Figure S1). Specifically, positive control plasma with AAV5 NAb inhibited liver FVIII expression by 53.8% (P=0.14 vs negative control plasma) in the in vivo TI assay. The same positive control plasma sample resulted in titers of 1:7 070 and >1:60 000 in the in vitro cell-based AAV5 TI and AAV5 TAb assays, respectively. The variability and insufficient sensitivity of the in vivo mouse AAV5 TI assay precluded us from selecting this format for additional analytical validation. In light of these data, and the successful use of an AAV5 TAb assay and an in vitro AAV5 TI assay to select NHP subjects for enrollment, we chose to validate an AAV5 TAb assay and an in vitro cell-based TI assay as screening tools to help identify human patients for pre-existing immunity to AAV5. We developed a sequential bridging electrochemiluminescence assay (ECLA) to detect AAV5 antibodies in human plasma (Figure 2a), rather than an ELISA-based colorimetric assay,7 to lower the background signals detected (data not shown). The AAV5 TI cell-based format for human plasma was converted from a GFP-reporter system to a more high-throughput and sensitive method utilizing an AAV5 vector containing the luciferase reporter gene driven by a CMV promoter (AAV5-Luc). Human plasma samples were tested in serial dilutions to determine the TI titer. Forty-eight hours following transduction, cells were lysed and luminescence measured. The presence of AAV5 neutralizing factors interfere with the transduction process and decrease in luminescence when compared with control (Figure 2b).

Assay formats used to detect anti-adeno–associated virus 5 (AAV5) antibodies and neutralizing factors. (a) Total antibodies against AAV5 were measured in human plasma using a bridging electrochemiluminescence assay. AAV5 capsids are coated passively onto a plate, plasma samples are added after blocking, and AAV5-specific antibodies are detected using ruthenylated AAV5 capsids. Positive screening results are confirmed by adding excess unlabeled AAV5 capsids (not depicted). (b) The cell-based transduction inhibition assay measures the ability of plasma samples to reduce the transduction of 293T/17 cells by a recombinant AAV5 vector carrying a luciferase reporter. Luciferase-reported fluorescence might be reduced by interference with multiple steps, such as reduced interaction of AAV5 with its cognate cell surface receptors or interference with steps subsequent to the receptor-binding process.
Performance characteristics during analytical validation
Cut point determination
AAV5 TAb screening cut point and titer cut point: To determine the screening cut point (SCP) and titer cut point (TCP), 48 human plasma samples from healthy individuals were assessed in six independent experiments using the AAV5 TAb assay. The mean of duplicate AAV5 TAb assay values was normalized to the cut point control (CC) (negative control plasma) values. Zero of six experiments showed normally distributed data (P<0.05; Supplementary Figure S2a). Inclusion of samples containing AAV5 antibodies (biological outliers) and samples with unusually high signals (statistical outliers) in cut point analysis would likely inflate the cut point, which could contribute to a higher false-negative rate. Therefore, we excluded the outliers as described in the Materials and Methods section from the evaluation of the cut points. After outlier exclusion, the data from 4 of 6 runs were normally distributed (Supplementary Figure S2b). The data used in the SCP/TCP analysis are graphed with the specificity cut point data in Figure 3a. Means of normalized ELCU were significantly different across experiments (analysis of variance F test, P<0.05), and the variances were similar (Levene’s test, P⩾0.05) (Supplementary Figure S2c); therefore, a floating cut point calculation was applied.32 A parametric approach was used to calculate the SCPs and TCPs based on the 95th and 99th percentile upper limits, respectively. The SCPs and TCPs were determined to be 1.15 and 1.30, respectively.

Determination of adeno-associated virus 5 (AAV5) total anti-capsid antibody (TAb) and transduction inhibition (TI) assay cut points and sensitivities. (a) Assay cut point data for the AAV5 TAb assay. Samples from 48 healthy individual donors were tested in six experiments. y axis: normalized screening results (sample electrochemiluminescence unit [ECLU] normalized to ECLU of cut point control [CC]); x axis: percentage signal inhibition in the confirmatory/specificity step. The horizontal line indicates the screening assay cut point of 1.15. The vertical dashed line indicates the specificity cut point of 39.7% signal inhibition. (b) Assay cut point data for the AAV5 TI assay. Samples from 100 healthy individual donors were tested in six experiments. y axis: percentage transduction (sample relative light unit [RLU] normalized to RLU of CC pool) with the data points from one individual tested in one experiment. The horizontal line indicates the assay cut point of 44.9% transduction. Samples in (a) and (b) that showed an intraduplicate CV>25% in a single cut point run were removed from the analysis (red). Biological (blue) and statistical (green) outliers were also removed from the analysis (refer to the Materials and Methods section for outlier removal criteria). The remaining sample results (black) were used to derive statistical cut points. (c) Sensitivity for the AAV5 TAb assay. Six independent 1:5 serial dilutions of a polyclonal positive control anti-AAV5 antibody in pooled human plasma were analyzed. Horizontal line: assay screening cut point of 1.15 was used to determine a limit-of-detection (LOD) of 4.9 ng ml−1 in plasma. (d) Sensitivity for the AAV5 TI assay. Sixty-nine independent 1:2 serial dilutions of a monoclonal positive control anti-AAV5 antibody were run. Horizontal line: assay cut point of 44.9% transduction was used to determine an average LOD of 65.9 ng ml−1 in plasma. In c and d, the LOD was calculated for each curve as the concentration of control antibody that crosses the assay cut point. Only the data points that screened above and below the respective assay cut points were used in the sensitivity calculations.
AAV5 TAb specificity cut point: To determine the specificity cut point for the AAV5 TAb assay, the percentage signal inhibition (%SI) was calculated following competition of AAV5 antibodies by adding unlabeled AAV5 capsid. Forty-eight human plasma samples from healthy individuals were assessed in six independent experiments using the AAV5 TAb assay before and after competition. The distribution of each experiment was evaluated for normality using the Shapiro–Wilk test, with zero out of six experiments being normally distributed (P<0.05) (Supplementary Figure S3a). Biological and statistical outliers were identified as described in the Materials and Methods section and were excluded from the cut point evaluation. After removal of outliers, four of six runs were normally distributed (Supplementary Figure S3b). The data used in the specificity cut point analysis are graphed with the screening data in Figure 3a. Variances were pooled to calculate a fixed, parametric specificity cut point (using a 99% upper limit) at 39.7% SI.
AAV5 TI assay cut point: Because samples are simultaneously screened and titered in the AAV5 TI assay, we established only one assay cut point (ACP) that was used for both screening and titer assessment of samples. To determine the assay cut point, 100 plasma samples from healthy individuals were tested in six independent experiments using the AAV5 TI titer assay. All the assay data were normalized to the plate CC sample; no additional data transformations were performed for the final cut point analysis. To establish a conservative cut point and minimize the possibility of false-negative results, outliers were removed as described in the Materials and Methods section. None of the six experiments were normally distributed (Supplementary Figure S4), and log transformation of data did not result in normal distribution (data not shown). Therefore, the assay cut point was determined using a nonparametric method as the 99th percentile of the data set, at 44.9% transduction.
Determination of assay sensitivity
Assay sensitivities for the AAV5 TAb and TI assays were determined as the concentrations at which signal from positive control spiked samples intersected the assay SCP and ACP, respectively. Positive control samples were prepared by spiking a known amount of AAV5-specific antibody into human plasma.
AAV5 TAb sensitivity (limit of detection, LOD): To determine the AAV5 TAb assay sensitivity, six independently prepared positive control dose–response curves were generated by serial dilution of an anti-AAV5 rabbit polyclonal positive control antibody in pooled CC plasma. Using the two data points immediately above and below the assay SCP of 1.15 electrochemiluminescence unit (ECLU)/CC, a linear regression model extrapolated the point of intersection (Figure 3c). The mean sensitivity was 4.9 ng ml−1.
AAV5 TI sensitivity (LOD): To determine the sensitivity of the AAV5 TI assay, 69 independently prepared positive control dose–response curves were generated by serial dilution of a mouse monoclonal positive control anti-AAV5 antibody (ADK5b) in pooled CC plasma. Using the two data points above and below the assay TCP of 44.9% transduction, a linear regression model extrapolated the point of intersection (Figure 3d). The mean TI assay sensitivity was 65.9 ng/ml.
To provide better comparability between the sensitivities of the TAb and TI assay, the sensitivity of the TAb assay was also evaluated by calibrating the rabbit polyclonal anti-AAV5 serum (used in the previous section) against a mouse monoclonal anti-AAV5 antibody (ADK5a). ADK5a has a similar neutralizing potential as ADK5b in the TI assay (data not shown). The sensitivity of the TAb assay, when expressed as ‘ADK5a equivalents’ would be 68.9 ng ml−1 (Supplementary Figure S5), which is similar to the sensitivity obtained using ADK5b in TI assay.
Determination of assay specificity
AAV5 TAb assay specificity: The specificity of the TAb assay for AAV5 antibodies was demonstrated by two approaches. First, anti-AAV8 monoclonal antibodies were added to CC pooled plasma at concentrations up to 4 μg ml−1 (the specificity of AAV8 antibodies is shown in Supplementary Figure S6). These samples screened below the SCP for every concentration of anti-AAV8 antibodies, indicating that the assay does not cross-react with anti-AAV8 antibodies (Figure 4a). Second, plasma with anti-AAV5 antibodies added at either the high-quality control (HQC) or low-quality control (LQC) concentration were incubated with unlabeled AAV5 capsid added at concentrations from 0 to 2.0 × 1012 vg ml−1 (Figure 4b). A capsid dose-dependent decrease in signal was observed for both quality control (QC) concentrations, indicating the specificity of this assay for AAV5 capsid.

Evaluation of adeno-associated virus 5 (AAV5) total antibody (TAb) and transduction inhibition (TI) assay specificity. (a+b) Assay specificity of the AAV5 TAb assay. (a) Specificity of the anti-AAV5 TAb assay: increasing concentrations of a mouse anti-AAV8 antibody were spiked into pooled plasma. Unspiked samples were analyzed in triplicate, spiked samples in duplicate. Pooled plasma spiked with anti-AAV8 had a normalized electrochemiluminescence unit (ECLU) below the screening cut point (SCP) of 1.15. Low-quality control (LQC) sample spiked with 9 ng ml−1 of anti-AAV5 polyclonal antibody showed a normalized value above the SCP. (b) AAV5 specificity was further assessed in the TAb assay using spiked controls (LQC, dark gray bars; high-quality control [HQC], light gray bars) incubated with increasing concentrations of unlabeled AAV5 capsid. (c, d) Assay specificity for the AAV TI assay. (c) Specificity for the AAV5 TI assay: increasing concentrations of an anti-AAV8 monoclonal antibody were spiked into pooled plasma and added to AAV5-luciferase before being incubated with human embryonic kidney (HEK) 293T/17 cells. CC and LQC plasma samples spiked with 180 ng ml−1 of monoclonal AAV5 antibody are shown. Horizontal line: TI assay cut point at 44.9% transduction. (d) Competition with AAV5-Factor VIII (FVIII) to further demonstrate AAV5 TI assay specificity: AAV5-luciferase reporter vector was co-incubated with increasing concentrations of the AAV5 drug product before being added to HEK293T/17 cells. Horizontal line: TI assay cut point at 44.9% transduction. All the data (a–d) are represented as means±s.d.
AAV5 TI assay specificity: Specificity of the AAV5 TI assay for AAV5 was demonstrated by two approaches. First, anti-AAV8 monoclonal antibodies were added into pooled plasma and incubated with AAV5-luciferase prior to the AAV5 TI assay. All samples screened above the assay cut point (44.9%), and at ≈100% transduction levels, indicating that up to 48 μg ml−1 of this anti-AAV8 antibody did not interfere with the transduction efficiency of AAV5-luciferase (Figure 4c). Second, AAV5-luciferase was incubated with increasing concentrations of AAV5-FVIII prior to the assay. Concentration-dependent inhibition of luciferase transduction was observed with increasing concentrations of AAV5-FVIII (Figure 4d), indicating specific competition with AAV5-luciferase uptake.
Assay selectivity and matrix interference
AAV5 TAb assay selectivity and matrix interference: To assess potential interference from plasma components (that is, matrix interference) in the AAV5 TAb assay, samples from 10 healthy donors, six lipemic donors, and six hemolytic donors were assessed with the TAb screening assay, with or without addition of anti-AAV5 at the LQC concentration of 9 ng ml−1. Eight of 10 samples from healthy individuals without added anti-AAV5 antibody screened negative (Figure 5a), as did samples from four of six individuals with lipemia and from all six hemolytic donors (Figure 5b). All samples spiked with anti-AAV5 antibody screened positive. These results passed the acceptance criteria (Supplementary Table S1) set for the selectivity experiments recommended in regulatory guidance and indicate that lipemia and hemolysis do not interfere in the AAV5 TAb assay.

Evaluation of adeno-associated virus 5 (AAV5) total antibody (TAb) and transduction inhibition (TI) assay selectivity and matrix interference. (a) Ten healthy donors (M1–10). (b) Six lipemic donors (Lip M1–6) and six hemolytic donors (Hem M1–6). All samples were spiked with 9 ng/ml of an anti-AAV5 antibody (gray bars) or left unspiked (white bars) and tested in the anti-AAV5 TAb assay. Horizontal lines: TAb screening cut point of 1.15. (c) Ten healthy donors (M1–10) and 10 donors with hemophilia A (HA M1–10). (d) Nine lipemic donors (Lip M1–9) and nine hemolytic donors (Hem M1–9). All samples were spiked with 180 ng/ml of an anti-AAV5 antibody (gray bars), or left unspiked (white bars) and tested in the AAV5 TI assay. Horizontal lines: TI cut point of 44.9% transduction. All the data (a–d) are represented as means±s.d.
AAV5 TI assay selectivity and matrix interference: To assess potential matrix interference in the AAV5 TI assay, samples from 10 healthy donors, 10 donors with HA, nine lipemic donors, and nine hemolytic donors were assessed with the AAV5 TI assay, either with or without addition of anti-AAV5 antibody at the LQC concentration of 180 ng ml−1. For the samples from healthy individuals and individuals with HA, the unspiked samples showed no detectable TI activity, because values for all screened samples were above the assay cut point (Figure 5c). Spiked samples showed transduction levels ranging from 0.5 to 31.3%, which were below the assay cut point. In unspiked lipemic samples, six of nine showed TI by screening below the assay cut point (Figure 5d), as did all spiked samples. In unspiked hemolytic samples, three of nine samples screened positive, as did all spiked samples. These results indicate that hemolysis and lipemia could interfere with the AAV5 TI assay. However, definitive evidence of interference could not be obtained, because it was not possible to determine whether the tested hemolytic and lipemic samples also contained other pre-existing neutralizing factors that could explain the decrease in transduction.
AAV5 TAb assay precision: To assess inter- and intra-assay precision, two sets of QCs (at concentrations of HQC, LQC, and negative QC [(NQC]) were run on 30 plates over 10 days (three plates per day) by two analysts, and the coefficient of variation (CV) was determined. All analyses were performed on data normalized to the CC pool. Interassay precision values for HQC, LQC, and NQC were 14.3, 13.1, and 9.9% CV, respectively. Intra-assay precision values for HQC, LQC, and NQC were 4.6, 7.2, and 6.3% CV, respectively. Titer QC (TQC) precision was also assessed. Analyses were based on the same six curves used to determine assay sensitivity. Precision for TQC was 5.5% CV.
AAV5 TI assay precision: To assess inter- and intra-assay precision, two or three sets of QCs for the LQC and NQC (n=125) were run on each of 80 plates over 10 days (eight plates per day) by two analysts. All analyses were performed on data normalized to the CC pool. Interassay precision values for LQC and NQC were 17.5 and 35.6% CV, respectively. Intra-assay precision values for LQC and NQC were 11.6 and 13.2% CV, respectively. TQC precision analyses were based on the same 69 curves used to determine assay sensitivity. TQC precision was 3.8% CV. A summary of the assay validation parameters and performance characteristics are provided in Table 1.
Dual-assay screening suggests AAV5 transduction inhibition by non-antibody factors
We tested 124 human plasma samples (100 samples from healthy individuals and 24 from individuals with HA) in the AAV5 TAb and TI assays to evaluate cross-reactivity between the two assays. Assay data from healthy individuals are presented in Figure 6a and from HA individuals in Figure 6b. For healthy samples, the majority (53%, n=53) screened negative in both assays; 18% (n=18) of samples contained AAV5 NAb as evidenced by screening positive in both assays; 5% (n=5) of samples contained AAV5 antibodies that were not neutralizing based on positive results in the TAb assay but negative results in the TI assay. Last, 24% (n=24) of the healthy samples had non-AAV5 antibody-based neutralizing activities screening negative in the TAb assay but positive in the cell-based TI assay. Of the limited HA data set, the majority of samples screened negative in both assays (71%, n=17); 13% (n=3) of samples contained AAV5 NAb, screening positive in both assays; 8% (n=2) of samples had AAV5 antibodies that were not neutralizing; and 8% (n=2) of samples contained non-AAV5 antibody–based neutralizing factors. These data demonstrate that four subgroups exist in both healthy and HA populations. The percentages for each subgroup varied but TI positivity were not statistically different between the healthy and HA populations (Fisher’s exact test p=0.0636).

Dual-assay strategy identifies non-adeno-associated virus 5 (AAV5) antibody-based neutralizing factors in human plasma. Plasma samples from (a) 100 healthy donors (circles) and (b) 24 donors with hemophilia A (triangles) were tested in both validated AAV5 total antibody (TAb) and transduction inhibition (TI) assay. Sample data are represented as interpolated titer values. Both data sets exhibit four distinct groups: (1) samples that screened negative in both assays (circled in green), (2) samples that screened positive in both assays (circled in red), (3) samples that screened positive in the TAb assay and negative in the TI assay (circled in yellow), and (4) samples that screened negative in the TAb assay but positive in the TI assay (circled in blue). (c) Percentage of each group and number of samples in each group (boldface and italic) are presented, segregated by status: healthy, hemophilia A, and combined.
Putative non-antibody-based neutralizing factors in human plasma remain after immunoglobulin depletion
Next, we investigated in more detail whether the AAV5 TI activities in the human samples were exclusively mediated by antibodies. Samples that tested positive in the cell-based TI assay that were either positive (TI+/TAb+) or negative (TI+/TAb−) in the AAV5 TAb assay were depleted of immunoglobulins (Igs) using protein A/G/L columns. To ensure that antibodies were successfully removed by protein A/G/L column treatment, LQC (180 ng ml−1) and TQC (3000 ng ml−1) samples spiked with anti-AAV5 antibodies were similarly depleted of Igs using protein A/G/L columns. The depleted samples were then tested in the AAV5 TI assay. LQC and TQC samples depleted with protein A/G/L treatment showed % transduction levels similar to those of CC negative control, indicating successful removal of antibodies. After Ig depletion, nine of 10 AAV5 TI+/TAb+ samples showed a level of transduction above the TI assay cut point after Ig depletion, confirming that these samples contained NAb against AAV5, while one of 10 AAV5 TI+/TAb+ samples continued to show TI after immunodepletion. Conversely, 12 of 20 AAV5 TI+/TAb− samples that were Ig-depleted showed levels of transduction above the assay cut point, whereas the remaining eight samples still showed % transduction below the assay cut point in the cell-based TI assay, suggesting that antibodies are unlikely to be the neutralizing factors in these samples.
Discussion
In this study, we analytically validated AAV5 TAb and cell-based TI assays and determined assay cut points using a conservative statistical approach for the identification of individuals who lack pre-existing TAb or neutralizing factors to AAV5. Using these two assays to evaluate procured human plasma samples, we showed that a majority of the samples were cross-reactive in both assays (that is, screened positive or negative in both assays). However, there was a subgroup of both NHP and human samples (including normal and HA) that were only positive in the cell-based transduction inhibition assay. The presence of unidentified factors in serum that might be responsible for AAV neutralization was first suggested in Boutin et al.7 Non-clinical experiments to determine the impact of these non-antibody neutralizing factors on AAV transduction efficiency in vivo can be conducted by dosing TI-positive but TAb-negative animals.
Efficacy, safety, and ethical considerations are the primary reasons to consider enrolling patients who have acceptable levels of AAV pre-existing immunity, which needs to be investigated and confirmed clinically. The relevance of testing for NAb was previously demonstrated in a study in which AAV8 gene therapy vector was administered to macaques with different levels of pre-existing AAV8 NAb, leading to the suggestion that subjects with NAb titers >1:10 (as measured by an in vitro TI assay) should be excluded from AAV8 gene therapy trials,33 although it is important to note that levels of titers measured in one assay cannot be used in other assays because titer determination is dependent on assay sensitivity. The studies reported in Nathwani et al1 and Jaski et al34 enrolled patients using assays measuring TI in vivo and in cells, respectively.1, 16, 27, 30, 31 Similarly, the presence of antibodies (neutralizing or not) against AAV5 vector needs to be considered, because subjects with prior exposure to wild-type AAV5 might mount a recall humoral response and an adaptive cell-mediated response following AAV5 vector administration.6, 34 It is conceivable that pre-existing and recall anti-AAV5 antibodies might prevent efficient uptake of gene therapy vector or provoke a cytotoxic T-cell response that might result in loss of transduced cells, affecting efficacy or safety outcomes.4, 34, 35 Furthermore, because of the impact of the expected host humoral immune response to AAV capsid, patients may currently only have one chance of receiving AAV5-based gene therapy. Therefore, we reason that a conservative screening approach to enroll patients that are negative in both assays could increase the likelihood of achieving proof-of-concept in early phase 1/2 trials of AAV5-based gene therapy.
To detect neutralizing factors, the cell-based TI assay uses human embryonic kidney (HEK) 293T/17 cells as the host to measure transduction of AAV5 vector containing a cytomegalovirus (CMV)-driven luciferase construct. This cell line was used in previous studies for NAb detection and patient screening.12, 13, 16 During the TI assay development phase, we tested several other cell lines in addition to HEK293T/17 cells, including HepG2 and Huh7. However, we found that a much higher multiplicity of infection (MOI) was needed to transduce HepG2 or Huh7 cells, rendering the TI assay less sensitive in these cells (data not shown) as compared with an MOI of 25 000 to transduce HEK293T/17 cells. Others have also shown that many parameters can influence the sensitivity of the cell-based TI assay, including the choice of cell line and positive control antibodies, cell growth condition, MOI, and the empty to full capsid ratio.17, 28 Therefore, it is important to standardize these parameters and secure sustainable supplies of the reagents in order to ensure reproducibility and track assay performance.
To ensure accurate and reproducible detection of AAV5 immunity in human samples, we analytically validated the TAb and TI assays in adherence to industry guidelines for immunogenicity assays.32 In contrast with previously published AAV-based screening methods for which arbitrary thresholds were used to determine positivity and titers,1, 7, 16, 27 we used a conservative statistical approach32 to determine assay cut points and minimize false-negative results. A caveat of the approach taken here is that the cut points for both assays were determined using samples from healthy male donors because of the limited availability of samples from patients with severe HA. These assay cut points will require additional confirmation in the patient population as larger sets of HA samples sourced from the relevant geographic locations become available.32
To understand the concordance of the TAb and TI assays, screening of 100 samples from healthy individuals and 24 samples from individuals with HA showed that the majority of samples cross-react in both assays (> 70%, Figure 6c). We identified a minority of samples with discordant assay results, the most interesting of which had neutralizing factors detected by the AAV5 TI assay but no anti-AAV5 antibodies detected by the TAb assay. Within this category of AAV5 TI-positive and AAV5 TAb-negative samples, some samples contained transduction neutralizing activity that could be removed with protein A/G/L treatment (Figure 7). These samples might contain antibodies directed against other proteins that are important for AAV5 transduction, such as the platelet-derived growth factor receptor or the recently identified AAV receptor.36, 37, 38, 39 We cannot rule out the possibility that these samples might contain antibodies directed against other AAV serotypes that are capable of binding to AAV5-luciferase capsids in solution but that were not detected using the plate-based AAV5 TAb assay described here. Other AAV5 TI-positive and AAV5 TAb-negative samples contain an AAV5 transduction neutralizing activity that cannot be removed with protein A/G/L treatment (Figure 7), suggesting that the factor is not an Ig. The nature of the factor(s) remains to be determined. Previous studies have demonstrated that many small molecules can modulate various steps of the AAV transduction process, including binding to cell surface receptors, internalization, intracellular transport, endosomal escape, nuclear transport, and gene transcription and translation using both in vitro and in vivo TI experimental models.19, 21, 23, 24, 25, 26 It remains an open question whether such neutralizing factors are present in human plasma samples arising from genetic, dietary, or environmental influence. Of interest, some drugs approved by the Food and Drug Administration that increase intracellular calcium influx (for example, paclitaxel, vinblastine, and heparin) have been shown to inhibit AAV-mediated transduction in assay systems.19, 20, 21, 23, 24, 25 Whether the AAV transduction process can be pharmacologically modulated in vivo warrants further study. In addition to small molecules, factors in the bronchial secretion of patients with cystic fibrosis, likely neutrophil peptides, have also been shown to substantially inhibit in vitro AAV transduction.22 An alternative possibility is that some plasma may contain agents capable of interfering with aspects of the TI assay independent of AAV5 transduction; for example, steroids could regulate CMV promoter–directed gene expression,40 which might not be relevant for AAV vectors using different promoters. In addition, we cannot rule out the possibility of cytotoxic factors present in the plasma samples, although cell numbers and health were visually confirmed. Nonetheless, while the nature of the neutralizing factors in these human plasma samples is not understood, our work highlights the importance of studying their biological relevance in vivo for AAV-based gene therapy.

Non-antibody-mediated non-adeno-associated virus 5 (AAV5) transduction inhibition (TI) neutralizing factors are present in human plasma. Thirty human plasma samples that tested positive in the TI assay were subjected to immunodepletion on agarose protein A/G and protein L resins and tested in the AAV5 TI assay. The percentage transduction values before (white bars, controls) and after (black bars) depletion are shown. All the data are represented as means±s.d.s. The assay cut point is represented by the black horizontal line. CC, cut point control; LQC, low-quality control; TQC, titer quality control.
The prevalence of AAV5 immunity has been studied in healthy donors, patients with cystic fibrosis, and pediatric patients with HA.7, 9, 10, 11, 12, 41 However, the prevalence of AAV5 immunity in adults with severe HA is largely unknown. The prevalence of different AAV serotypes has been shown to vary with geographic location and age.8, 10, 12 Several groups reported the highest neutralizing factor seroprevalences were observed for AAV2 (10–60%) and AAV1 (3–67.7%), while the lowest were observed for AAV8 (1–19%) and AAV5 (3.2–9%) in adult populations.7, 10, 11, 16 Furthermore, most of these studies focus on the prevalence of NAb against different serotypes of AAV and potential cross-reactivity from antibodies against AAV2. We propose that the two assays described herein may serve as platforms to study the prevalence and the relationship between TAb and TI in the intended patient population.
Materials and methods
Plasma samples
All plasma samples were collected using sodium citrate as anticoagulant. All individuals and pooled human plasma samples were purchased from Bioreclamation (Hicksville, NY, USA), except for six samples from HA donors that were purchased from George King Biomedical Inc (Overland Park, KS, USA). The 100 healthy male donors varied in ethnicity (black, white, Hispanic, and Asian) and ages (20–63 years). The 24 samples from male donors with HA were of black and white ethnicities aged 21–68 years. The CC used in the human TI assay was pooled from 16 healthy individual donors with low (<10% TI) to no TI. The CC plasma pools used in both human and NHP TAb assays and in the NHP TI assay were purchased from Bioreclamation. Three human male plasma pools were tested in the human TAb assay, and the pool generating low background signals was selected as CC control (data not shown). Plasma samples indicating hemolysis were from individual healthy donors with high (≈1100 mg dl−1), middle (≈275 mg dl−1), and low (≈35 mg dl−1) levels of hemoglobin. Plasma samples indicating lipemia were from individual healthy donors and assigned high, middle, and low levels by the vendor.
Antibody reagents
The antibody used as a positive control in the TAb assays and the NHP TI assay was a rabbit polyclonal antibody serum (catalog no. 20R-2587; Fitzgerald Industries International, Acton, MA, USA) that specifically reacts with capsid proteins VP1, VP2, and VP3 of AAV5 and has no cross-reactivity with AAV types 1 to 4. The positive control used in the human TI assay was a mouse monoclonal antibody (clone ADK5b) from LifeSpan Biosciences (Seattle, WA, USA) that specifically binds intact AAV5 particles by recognizing a conformational epitope on assembled capsids that is not present in denatured or unassembled capsid proteins, according to vendor information. The mouse monoclonal anti-AAV5 antibody (ADK5a) used to calibrate the rabbit polyclonal anti-AAV5 serum was from Progen Biotechnik GmbH (Heidelberg, Germany). The anti-AAV8 antibody used for specificity testing was a mouse monoclonal antibody (clone ADK8) specific for intact AAV8 capsids (Fitzgerald Industries International), with no cross-reactivity to AAV types 1 to 6 and 9, according to vendor information.
AAV reagents
The AAV5 capsid used in the NHP TAb assay was AAV5-FVIII produced in insect Sf9 cells and formulated in a buffer containing 0.001% pluronic F-68. The AAV5-FVIII was not modified for coating plates or in the specificity assay. For detection in the human TAb assay, AAV5-FVIII was buffer-exchanged to Dulbecco’s phosphate-buffered saline (DPBS) without calcium and magnesium supplemented with 0.2% Pluronic F68 (Alfa Aesar, Ward Hill, MA, USA) using three cycles of filtration with Amicon-15 centrifugal filters (Millipore, Billerica, MA, USA). Next, the buffer-exchanged AAV5-FVIII was labeled with 10 nmol MSD Sulfo-NHS Ruthenium (Rockville, MD, USA) for 1 h at ambient temperature with rotation. The ruthenium-labeled AAV5-FVIII was purified via three additional cycles of filtration with Amicon-15 centrifugal filters to phosphate-buffered saline (PBS) with 0.2% Pluronic F68 and stored at −80 °C in single-use aliquots. The unmodified AAV5-FVIII was also used for specificity experiments in the human TI assay and in vivo TI assay. The NHP cell-based TI assay used AAV5-CMV-GFP purchased from SignaGen (Rockville, MD, USA). AAV5 vector used for transduction in the human TI assay was AAV5-CMV-Luciferase produced in insect Sf9.
General reagents
Unlabeled protein A/G/L used for detection in the NHP TAb assay was purchased from BioVision (Milpitas, CA, USA). The ruthenium-labeled protein A/G/L reagent was prepared by reconstituting protein A/G/L according to manufacturer’s recommendation at 10 mg/ml in DPBS without calcium and magnesium. The concentration was adjusted to 1 mg ml−1 in DPBS and labeled with 10 nmol MSD Sulfo-NHS Ruthenium for 1 h at ambient temperature with rotation. The labeled protein A/G/L was purified by three cycles of filtration with Amicon-15 centrifugal filters using DPBS and stored at −80 °C in single-use aliquots. Tris Buffered Saline with 1% (w/v) Casein (TBS-C) was from BioRad (Hercules, CA, USA). The protein A/G columns, protein L columns, and bicinchoninic acid (BCA) protein assay kits were from Thermo Fisher Scientific (South San Francisco, CA, USA). Etoposide was from Enzo Life Sciences (Farmingdale, NY, USA), Steady-Glo Luciferase reagent was from Promega (Sunnyvale, CA, USA), fetal bovine serum (FBS) without heat inactivation was from HyClone (Logan, UT, USA), and Penicillin-Streptomycin-Glutamine (100 ×) was from Gibco (South San Francisco, CA, USA). The HEK293T/17 cell line was purchased from ATCC (Manassas, VA, USA). The GFP quantification kit was purchased from Cell Biolabs (San Diego, CA, USA).
AAV5 TAb assay for nonhuman primate plasma
TAb against AAV5 were detected in NHP plasma using a sandwich ECLA on the MSD platform. All plate incubation steps were performed for 1 h with shaking at ambient temperature, followed by washing with TBST (DBPS, 0.1% Tween 20, 0.05% Proclin300). First, bare standard-bind Multi-Array MSD plates were coated with 5.0 × 1011 vg ml−1 AAV5 in PBS and blocked with TBS-C (1% casein in 20 mM Tris, 500 mM NaCl; pH 7.4). QCs were prepared by diluting a rabbit polyclonal anti-AAV5 antibody 1:10 in 100% NHP pooled plasma and then subsequently by a factor of three for a total of eight dilutions. Pooled plasma without antibody was also tested as a negative CC. QCs and test samples were diluted at the minimum required dilution (MRD) of 1:20 in TBS-C and added in duplicate to the plate. For detection, 0.1 μg ml−1 ruthenium-labeled protein A/G/L in TBS-C was added, electrochemiluminescence was detected by adding 1 × MSD Read Buffer containing the substrate tripropylamine by the MSD QuickPlex using Discovery Workbench software version 4.0.12, and the signal was expressed in relative ECL units. Sample results were reported as a signal-to-noise (S/N) value, calculated by dividing sample ECL units by CC ECL units.
AAV5 TAb assay for human plasma
TAb against AAV5 was measured in human plasma using a sequential bridging ECLA on the MSD platform. All plate incubation steps were performed for 1 h with shaking at ambient temperature, followed by washing with TBST. Standard-bind Multi-Array MSD plates were coated with 2.16 × 1012 capsids per ml AAV5 in PBS and blocked with TBS-C. QCs were prepared in 100% pooled human CC plasma using a rabbit polyclonal anti-AAV5 antibody at 9 ng ml−1 (LQC) and 1000 ng ml−1 (HQC). Pooled plasma without antibody was also tested as a negative control CC. QCs and test samples were diluted at the MRD of 1:20 in TBS-C (screening assay) or in TBS-C containing 1.08 × 1012 capsid per ml AAV5 (specificity/confirmatory assay) and added in duplicate to the plate. In the titer assay, MRD samples were serially diluted 1:5 and tested as in the screening assay. Serially diluted HQC served as a TQC. For detection, 1.0 μg ml−1 ruthenium-labeled AAV5 capsid in TBS-C was added, and after the addition of 2 × MSD Read Buffer T, electrochemiluminescence was detected by the MSD Sector Imager 2400 using Discovery Workbench software version 3.0.18. Sample results were expressed as an S/N value, calculated by dividing sample ECL units by CC ECL units. For the specificity/confirmatory assay, 1 minus the ratio of the AAV5 capsid-treated sample S/N value divided by the untreated sample S/N value was expressed as percentage depletion.
AAV5 TI assay for nonhuman primate plasma
The AAV5 TI assay for NHP plasma used HEK293T/17 seeded at 2 × 106 cells per well in 60-mm tissue culture dishes in Dulbecco’s modified eagle medium (DMEM) containing 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, and 2.92 mg/ml L-glutamine. On the next day, the LQC, MQC, and HQC were prepared with a rabbit polyclonal antibody in heat-inactivated NHP pooled plasma at concentrations of 0.5, 1.0, and 5.0 μg ml−1, respectively. QCs and samples were either treated with protein A/G for depletion (specificity assay) or left untreated (screening assay) and then diluted to MRD 1:20 in a mixture containing AAV5-CMV-GFP vector at 20 000 MOI in serum-free DMEM and incubated for 1 h at 37 °C. One wash was performed with serum-free DMEM and samples were added to the plate in duplicate. After 1 h, 3 volumes of DMEM containing 10% FBS was added. The infection was allowed to progress for 3 days with the media refreshed at least once over the course of the incubation. Cells were lysed with 0.5 ml of 1 × cell lysis solution supplied with the GFP quantification kit and shaken for 10 min at room temperature. Lysates were transferred to microcentrifuge tubes and centrifuged to remove any cellular debris. Supernatants were collected and separated into two equal aliquots. Lysates were stored at −80 °C until analyzed for GFP concentration using a GFP quantification kit and total protein concentration using a BCA protein assay kit. The amount of GFP was normalized to the total protein content. Data were expressed as percentage transduction calculated as normalized GFP of plasma samples divided by normalized GFP of negative control plasma.
Cell-based AAV5 TI titer assay for human plasma
The TI assay for human plasma samples used HEK293T/17 cells from a working cell bank generated with 5 million cells per vial. Each vial was single use and was not continuously passaged. Cells were thawed, counted, and seeded at 40 000 cells per well in warmed DMEM/10% FBS media in white clear-bottom 96 well plates avoiding all edge wells (60 wells per plate). On the next day, QCs prepared in 100% pooled human plasma (CC) using a mouse monoclonal anti-AAV5 antibody (ADK5b) at 180 ng/ml (LQC) and 3000 ng ml−1 (TQC) were thawed for use. TQC was titrated in seven serial 1:2 dilutions in CC. CC plasma without antibody was also tested as a negative control. QCs and test plasma samples were mixed 1:1 with 25 000 vg/cell MOI AAV5-CMV-Luciferase vector in DMEM with 1% bovine serum albumin for 30 min at ambient temperature before addition to cells in duplicate wells. After 1 h at 37 °C, the final concentration of 20 μM etoposide solution in DMEM 10% FBS was added. Cell confluency and health were monitored under the microscope daily and recorded if cytotoxicity was observed. After 2 days, media were removed and luciferase reagent was added for 10 min at ambient temperature with shaking. Luminescence was measured using a 500-ms integration time on a VICTOR microplate reader; software version 4.0. AAV5 TI titers were determined as the reciprocal dilution of plasma samples at the TCP.
Protein A/G/L depletion for the AAV5 TI assay for human plasma
Samples were first subjected to depletion using protein A/G columns and then protein L columns. All centrifugation steps, unless otherwise specified, were performed for 1 min at 5000 g at room temperature. Columns and buffers were equilibrated to room temperature. Columns were first spun to remove the storage buffer. The flow-through was discarded. Column equilibration was performed three times by adding 400 μl of binding buffer to the column/collection tube assembly. Columns were spun and the flow-through was discarded. A 125- μl of plasma sample was added to one column and subjected to end-over-end mixing for 10 min at room temperature. The columns were spun at 5000 g for 2 min, and flow-through was collected into a clean collection tube. The flow-through was subjected to additional depletion on protein L columns following the same procedure describe above. Final flow-through was assayed using the cell-based TI assay method.
References
Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 2014; 371: 1994–2004.
Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 2011; 365: 2357–2365.
Kay MA, Manno CS, Ragni MV, Larson PJ, Couto LB, McClelland A et al. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat Genet 2000; 24: 257–261.
Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 2006; 12: 342–347.
Genetic Modification Clinical Research System. Version 6.2. Available at: https://www.gemcris.od.nih.gov/Contents/GC_HOME.asp. Accessed 7 October 2016.
Masat E, Pavani G, Mingozzi F . Humoral immunity to AAV vectors in gene therapy: challenges and potential solutions. Discov Med 2013; 15: 379–389.
Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther 2010; 21: 704–712.
Calcedo R, Morizono H, Wang L, McCarter R, He J, Jones D et al. Adeno-associated virus antibody profiles in newborns, children, and adolescents. Clin Vaccine Immunol 2011; 18: 1586–1588.
Mingozzi F, Chen Y, Edmonson SC, Zhou S, Thurlings RM, Tak PP et al. Prevalence and pharmacological modulation of humoral immunity to AAV vectors in gene transfer to synovial tissue. Gene Ther 2013; 20: 417–424.
Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM . Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis 2009; 199: 381–390.
Halbert CL, Miller AD, McNamara S, Emerson J, Gibson RL, Ramsey B et al. Prevalence of neutralizing antibodies against adeno-associated virus (AAV) types 2, 5, and 6 in cystic fibrosis and normal populations: implications for gene therapy using AAV vectors. Hum Gene Ther 2006; 17: 440–447.
Li C, Narkbunnam N, Samulski RJ, Asokan A, Hu G, Jacobson LJ et al. Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene Ther 2012; 19: 288–294.
Moskalenko M, Chen L, van Roey M, Donahue BA, Snyder RO, McArthur JG et al. Epitope mapping of human anti-adeno-associated virus type 2 neutralizing antibodies: implications for gene therapy and virus structure. J Virol 2000; 74: 1761–1766.
Wang L, Calcedo R, Wang H, Bell P, Grant R, Vandenberghe LH et al. The pleiotropic effects of natural AAV infections on liver-directed gene transfer in macaques. Mol Ther 2010; 18: 126–134.
Grimm D, Lee JS, Wang L, Desai T, Akache B, Storm TA et al. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol 2008; 82: 5887–5911.
Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Pogoda JM et al. Prevalence of AAV1 neutralizing antibodies and consequences for a clinical trial of gene transfer for advanced heart failure. Gene Ther 2016; 23: 313–319.
Meliani A, Leborgne C, Triffault S, Jeanson-Leh L, Veron P, Mingozzi F . Determination of anti-adeno-associated virus vector neutralizing antibody titer with an in vitro reporter system. Hum Gene Ther Methods 2015; 26: 45–53.
Sun L, Tu L, Gao G, Sun X, Duan J, Lu Y . Assessment of a passive immunity mouse model to quantitatively analyze the impact of neutralizing antibodies on adeno-associated virus-mediated gene transfer. J Immunol Methods 2013; 387: 114–120.
Berry GM, Murlidharan G, Asokan A . Modulation of intracellular calcium enhances AAV transduction in the CNS. Mol Ther 2016; 24 (Suppl 1): S14.
Weinberg MS, Nicolson S, Bhatt AP, McLendon M, Li C, Samulski RJ . Recombinant adeno-associated virus utilizes cell-specific infectious entry mechanisms. J Virol 2014; 88: 12472–12484.
Hirosue S, Senn K, Clement N, Nonnenmacher M, Gigout L, Linden RM et al. Effect of inhibition of dynein function and microtubule-altering drugs on AAV2 transduction. Virology 2007; 367: 10–18.
Virella-Lowell I, Poirier A, Chesnut KA, Brantly M, Flotte TR . Inhibition of recombinant adeno-associated virus (rAAV) transduction by bronchial secretions from cystic fibrosis patients. Gene Ther 2000; 7: 1783–1789.
Mitchell AM, Samulski RJ . Mechanistic insights into the enhancement of adeno-associated virus transduction by proteasome inhibitors. J Virol 2013; 87: 13035–13041.
Mitchell AM, Li C, Samulski RJ . Arsenic trioxide stabilizes accumulations of adeno-associated virus virions at the perinuclear region, increasing transduction in vitro and in vivo. J Virol 2013; 87: 4571–4583.
Berry GE, Asokan A . Chemical modulation of endocytic sorting augments adeno-associated viral transduction. J Biol Chem 2016; 291: 939–947.
Nonnenmacher M, Weber T . Intracellular transport of recombinant adeno-associated virus vectors. Gene Ther 2012; 19: 649–658.
Nathwani AC, Rosales C, McIntosh J, Rastegarlari G, Nathwani D, Raj D et al. Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol Ther 2011; 19: 876–885.
Wang M, Crosby A, Hastie E, Samulski JJ, McPhee S, Joshua G et al. Prediction of adeno-associated virus neutralizing antibody activity for clinical application. Gene Ther 2015; 22: 984–992.
Davidoff AM, Gray JT, Ng CY, Zhang Y, Zhou J, Spence Y et al. Comparison of the ability of adeno-associated viral vectors pseudotyped with serotype 2, 5, and 8 capsid proteins to mediate efficient transduction of the liver in murine and nonhuman primate models. Mol Ther 2005; 11: 875–888.
Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 2011; 124: 304–313.
Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail 2009; 15: 171–181.
Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal 2008; 48: 1267–1281.
Wang L, Calcedo R, Bell P, Lin J, Grant RL, Siegel DL et al. Impact of pre-existing immunity on gene transfer to nonhuman primate liver with adeno-associated virus 8 vectors. Hum Gene Ther 2011; 22: 1389–1401.
Mingozzi F, High KA . Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood 2013; 122: 23–36.
Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med 2007; 13: 419–422.
Di Pasquale G, Davidson BL, Stein CS, Martins I, Scudiero D, Monks A et al. Identification of PDGFR as a receptor for AAV-5 transduction. Nat Med 2003; 9: 1306–1312.
Pillay S, Meyer NL, Puschnik AS, Davulcu O, Diep J, Ishikawa Y et al. An essential receptor for adeno-associated virus infection. Nature 2016; 530: 108–112.
Gurda BL, DiMattia MA, Miller EB, Bennett A, McKenna R, Weichert WS et al. Capsid antibodies to different adeno-associated virus serotypes bind common regions. J Virol 2013; 87: 9111–9124.
Harbison CE, Weichert WS, Gurda BL, Chiorini JA, Agbandje-McKenna M, Parrish CR . Examining the cross-reactivity and neutralization mechanisms of a panel of mAbs against adeno-associated virus serotypes 1 and 5. J Gen Virol 2012; 93: 347–355.
Malone RW, Hickman MA, Lehmann-Bruinsma K, Sih TR, Walzem R, Carlson DM et al. Dexamethasone enhancement of gene expression after direct hepatic DNA injection. J Biol Chem 1994; 269: 29903–29907.
Liu Q, Huang W, Zhang H, Wang Y, Zhao J, Song A et al. Neutralizing antibodies against AAV2, AAV5 and AAV8 in healthy and HIV-1-infected subjects in China: implications for gene therapy using AAV vectors. Gene Ther 2014; 21: 732–738.
Acknowledgements
We thank Drs Greg Hayes and Vivian Albert for helpful comments and Dr Kevin Ru for statistical analysis support. Editorial support was provided by Karl Zawadzki, PhD and M Evelyn Rose, PharmD, both of Health Interactions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
KS, BY, BL, BC, CV, SJZ and SF are employees of BioMarin Pharmaceutical, Inc., and ST is an employee of GenoSafe. During research and manuscript development, LF and LT were employees of BioMarin Pharmaceutical, Inc., and SG was an employee of Eurofin BioAnalytical Services.
Additional information
Supplementary Information accompanies this paper on Gene Therapy website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Falese, L., Sandza, K., Yates, B. et al. Strategy to detect pre-existing immunity to AAV gene therapy. Gene Ther 24, 768–778 (2017). https://doi.org/10.1038/gt.2017.95
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2017.95
This article is cited by
-
Adeno-associated virus as a delivery vector for gene therapy of human diseases
Signal Transduction and Targeted Therapy (2024)
-
Hybrid modeling of an ultracentrifugation process for separation of full and empty adeno-associated virus particles
Bioprocess and Biosystems Engineering (2024)
-
Clinical enrollment assay to detect preexisting neutralizing antibodies to AAV6 with demonstrated transgene expression in gene therapy trials
Gene Therapy (2023)
-
Validation of Anti-Adeno Associated Virus Serotype rh10 (AAVrh.10) Total and Neutralizing Antibody Immunogenicity Assays
Pharmaceutical Research (2023)
-
Immunogenicity of Recombinant Adeno-Associated Virus (AAV) Vectors for Gene Transfer
BioDrugs (2023)
