
Abstract
Glucokinase (GCK) is an important enzyme critical for glucose metabolism, and has been targeted as such in the pursuit of a cure for diabetes mellitus. We show that streptozotocin (STZ)-induced diabetic murine model exhibits low GCK expression with high blood glucose levels; moreover, aggravated glomerulonephritis is observed in the model when there is IL10 deficiency. Although T cells infiltrate into the liver and pancreas in STZ-induced diabetes mice, T helper 1 (Th1) and T helper 17 (Th17) cells decrease significantly with STZ addition in in vitro polarization. Using a mutant GCK gene (GCK 262) with a knocked out cytosine at position 2643 results in lower protein expression and more ubiquitination-led protein degradation compared with wild-type GCK (GCK 261). We further observed that hsa-mir-1302 can bind to 3′-untranslated region of mutant GCK, which can decrease GCK mRNA translation. Finally, delivery of mutant GCK by subcutaneous injection is more effective at decreasing blood glucose in the STZ-treated (STZ) murine diabetes model than insulin treatment alone. Similarly, mutant GCK consistently and moderately decreases blood glucose levels in GK rats over a period of 12 and 70 days without inducing hypoglycemia, whereas insulin is only effective over 12 h. These results suggest that mutant GCK may be a future cure for diabetes.
Similar content being viewed by others
Introduction
Diabetes is a chronic condition characterized by elevated levels of blood glucose, which may arise from a multitude of different reasons. Commonly classified into type 1 diabetes (T1D) or type 2 diabetes (T2D) based on age of onset, diabetes has been rising in prevalence around the world. A broad condition, T2D impacts a wide range of cell types, not all of which are directly related to metabolic function. Many different proteins have been identified to correlate with diabetes through genome-wide association studies, over a broad range of classes and cell types. For instance, the T-cell-specific1 HMG box transcription factor TCF7L2 is currently recognized as the strongest genetic marker for T2D.2 Similarly, β-cell expression of PTEN has been shown to be a contributing factor to T2D.3 In addition to those proteins, several others known to interact with insulin have been identified as being particularly important. Glucokinase (GCK) is one of these proteins that is expressed in many cells.4 A hexokinase that catalyzes the phosphorylation of glucose into glucose-6-phosphatase, GCK is commonly understood to be the first catalyst in the classical glucose metabolism pathway.5 GCK is also known for its ability to act as a sensor of glucose levels within cells. GCK exists in several distinct, but functionally similar, isoforms in different tissues, and its expression level varies accordingly. Many different mutations on the GCK gene have been linked to various types of diabetes, especially MODY and T2D.6
Many different therapies have been attempted to treat T2D, with several of them being focused on insulin. Metformin primarily acts to increase sensitivity to insulin,7 whereas sulfonylureas stimulate increased insulin secretion from β cells.8 Second-choice medications such as meglitinides9 and thiazolidinediones10 similarly target insulin. Recently, sodium-glucose cotransporter 2 inhibitors11 have been coming into use as an alternative therapy, by preventing the reabsorption of glucose by the kidneys, thereby allowing for elimination of excess glucose as waste. Bariatric surgery12, 13 has also been shown to restore blood glucose levels to normal ranges in many patients. However, all of these therapies have significant side effects, such as weight gain, or potential hypoglycemia, and these therapies are not always effective standing alone. As such, novel therapies for treating T2D are still clearly required.
In this paper, we use gene therapy to treat T2D, via a novel adenovirus-packaged mutant GCK gene. We used the gene in a number of in vitro and in vivo tests on different animal models of T2D, including GK rats and STZ-induced diabetic mice. We demonstrate that the mutant GCK gene alters the molecular machinery within cells through interaction with hsa-mir-1302, leading to lower and controlled levels of its expression. This controlled expression corresponds to a more gradual decrease in blood glucose levels. Through these results, we show that the mutant GCK gene is more effective than both insulin and the conventional GCK gene at ameliorating diabetes mellitus, and has the potential for stably controlling blood glucose levels over the long term.
Results
STZ reduces GCK expression in a murine model of diabetes
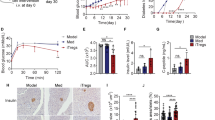
We first sought to confirm that GCK is indeed altered consistently in murine models of diabetes, especially the commonly used streptozotocin (STZ) model,14 as that model has been shown to be a useful mock of diabetes. STZ is known to preferentially target and kill the insulin-producing β cells of the islets of Langerhans because of their high expression of the GLUT2 (glucose transporter 2) transporter15 that STZ can co-opt. STZ was injected into B6 mice in 150 mg kg−1, leading to significant monotonic increases in the blood glucose levels of the mice after 3 days and lasting for 16 days postinjection (Figure 1a). The STZ-treated mice were of significantly lower weight on the day of killing (Figure 1b). As per our expectations, levels of GCK dropped significantly under STZ treatment on both protein (Figure 1c) and mRNA levels (Figure 1d) among liver cells. This result is consistent with previous reports from other groups.

STZ reduces GCK expression in a murine model of diatetes. The percentile changes of blood glucose (a) and body weight (b) of STZ-induced diabetic or PBS-treated mice. Data represent mean±s.d. (n=5), unpaired Student’s t-test, *P<0.05, and ***P<0.001. Western blot showed GCK protein expression (c) in liver homogenate of STZ-induced diabetic or PBS-treated mice. β-Actin expression was used as a control. Quantitative real-time RT–PCR (qPCR) (d) was performed for the analysis of mRNA expression of GCK treated as in (c). Data represent mean±s.d., unpaired Student’s t-test; ***P<0.001.
As other groups have previously demonstrated that T-cell tolerance of glucose has a major role in diabetes pathogenesis,16, 17 we next investigated the mice for T-cell-related alterations in their immune system. Interestingly, the effects of STZ in IL-10−/− mice were significantly more pronounced than in wild-type (WT) mice, in terms of both blood glucose (Supplementary Figure 1A) and body weight (Supplementary Figure 1B). GCK expression was also more greatly reduced as compared with WT-STZ-treated mice on the levels of both protein (Supplementary Figure 1C) and mRNA (Supplementary Figure 1D). Histological analysis also revealed the presence of greater amounts of inflammatory cells infiltrating into the kidney of the IL-10−/− mice as opposed to WT mice (Supplementary Figure 1E). These signs of glomerulonephritis are particularly significant as they have been implicated as part of a negative feedback loop that causes T2D to worsen in patients.18,19,20
Based on that result, we then further explored for a potential role being played by T cells in controlling GCK expression, given that IL-10 is understood to act strongly against T effector cell lineages. Immunofluorescent staining of the liver (Supplementary Figure 2A) and pancreas (Supplementary Figure 2B) slides revealed that significant amounts of CD3+ cells had infiltrated into the pancreas of STZ-treated mice. Flow cytometry of splenocytes isolated from WT mice with or without STZ-induced diabetes cultured in vitro in specific T-helper-polarizing conditions was then performed. Interleukin-17 (IL-17) secretion from Th17 group was clearly reduced in the STZ-treated group, with the overall population of IL-17-positive cells decreasing (Supplementary Figure 3A). The number of interferon-γ-positive Th1 cells was similarly lower under STZ treatment (Supplementary Figure 3B). However, less pronounced effects were observed with IL-4-positive T helper 2 (Th2) cells (Supplementary Figure 3C), suggesting that the effect of STZ may be somewhat selective. As none of the T-helper lineages are known to express the GLUT2 receptor that STZ may co-opt, it is unclear precisely what mechanism STZ is using to inhibit these cells.
We further applied the STZ model onto Rag−/− mice to clarify whether or not the expression of GCK was related to T- or B-cell activity. Blood glucose levels (Supplementary Figure 4A) were significantly increased with decreased body weight (Supplementary Figure 4B) in STZ-treated Rag-deficient mice compared with phosphate-buffered saline (PBS)-treated ones. Meanwhile, GCK protein expression (Supplementary Figure 4C) in the liver was reduced with a greater fold change in GCK mRNA expression (Supplementary Figure 4D). However, the pattern of blood sugar level change and body weight fluctuation was not significantly different from that observed in WT mice.
Mutant GCK has lower protein expression compared with WT GCK
Although we were unable to identify a clear correlation between GCK expression and a specific immune function, our results did confirm that GCK inhibition is linked to increased blood glucose levels. Although previous groups have identified that correlation and attempted to exploit it to treat diabetes via overexpression of GCK, they had difficulty in controlling the blood glucose levels with overexpression, and encountered hypoglycemia. As such, we instead attempted to devise a mutant copy of the GCK gene that would have more readily controllable expression (Supplementary Table 1). As a direct mutation in the protein itself may lead to unexpectedly detrimental side effects associated with exotic protein structures and potentially induce endoplasmic reticulum stress, we instead opted to attenuate the length of the 3′-untranslated region (UTR) by deleting a cytosine at base 2643. We consequently packaged the mutant GCK into a plasmid for experimentation.
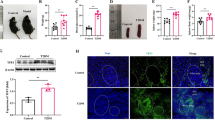
Because ubiquitination pathway has important role in protein degradation,21, 22 we transfected the mutant GCK plasmid into 293T cells, and it revealed significantly lower expression of GCK in cells transfected with mutant as compared with WT (Figures 2a and b). As such, we next sought to verify the molecular mechanisms that caused the lower expression of GCK in the mutant. We first cultured 293T cells overexpressing WT and mutant GCK, then ceased new mRNA synthesis with cycloheximide for 30 min and finally treated cells in the presence or absence of the proteinase inhibitor MG132. Treated mutant GCK degraded more than the untreated control over 12 and 24 h, whereas WT GCK showed stable expression (Figure 2c). Protein stability assays revealed that mutant and WT GCK had almost identical levels of ubiquitination (Figure 2d). Further transfections performed using specified lysine plasmids on ubiquitin suggests that K48 and K63 may not be particularly influential in GCK ubiquitination (Figure 2e). These results suggest that ubiquitination-independent pathway may involve in GCK protein degradation. Although it is possible that an alteration in the 3′-UTR may lead to changes in protein structure, such a possibility seems unlikely, given that the mutation in this case does not clearly impact the stop codon or otherwise cause a frameshift.

Mutant GCK has lower protein expression compared with WT GCK. (a) Green fluorescent protein (GFP)-tagged WT and mutant GCK plasmid were transfected into 293T cells for 36 h. Western blot was performed with anti-GCK. Green fluorescence was observed for both of GCK protein expression. (b) Enzyme assay analysis of WT and mutant GCK was overexpressed as in (a). (c) The 293T cells were transfected with WT and mutant GCK expression plasmid for 36 h. Under pretreatment with cycloheximide (10 μM) for 30 min, cells were then treated with or without MG132 for different time intervals. (d) The 293T cells were transfected with WT and mutant GCK overexpression plasmid and HA-tagged ubiquitin overexpression plasmid for 36 h. Then, cells were treated with or without MG132 for 12 h. The cell lysates were immunoprecipitated with anti-GCK antibody and immunoblotted with anti-HA antibody. (e) The 293T cells were transfected with WT and mutant GCK overexpression plasmid, HA-tagged ubiquitin overexpression plasmid, K48-only HA-tagged ubiquitin overexpression plasmid and K63-only HA-tagged ubiquitin overexpression plasmid for 36 h. The cell lysates were immunoprecipitated with anti-GCK antibody and immunoblotted with anti-HA antibody.
hsa-mir-1302 can bind to 3′-UTR of mutant GCK
We have observed mutant GCK degraded more in post-translational modification of ubiquitination pathway. As protein expression comprises of transcriptional and translational activities, we next probed for other phenomena that could potentially lead to the differences in protein expression observed. mRNA stability has long been identified as being a controlling factor of protein expression, and microRNAs have been found to potently impact mRNA activity.23, 24 As such, we next investigated to determine whether or not the deletion in the 3′-UTR had altered miRNA control of the GCK mRNA. Through sequence analysis,25 we identified the miRNA hsa-mir-1302 as being a potential binding partner to the mutant GCK 3′-UTR (Figure 3a). Although the binding of hsa-mir-1302 at the mutant 3′-UTR is not perfectly centered, previous groups have demonstrated that imperfectly centered sites were common, and may still effectively lead to mRNA repression.26 As such, we tested for the binding of the hsa-mir-1302 to the GCK 3′-UTR using a mimic. hsa-mir-1302 mimic did not affect the protein expression levels of WT GCK, but significantly reduced the expression of mutant GCK in a dose-dependent manner (Figure 3b). Similarly, addition of hsa-mir-1302 inhibitor did not affect the expression of WT GCK while leading to a slight increase in the expression of mutant GCK (Figure 3c). Taken together, hsa-mir-1302 reduces mutant GCK transcription activities without affecting WT ones.

Mutant GCK is a direct target of hsa-mir-1302. (a) Comparison of base pairs between hsa-mir-1302 and WT GCK or mutant GCK 3′-UTR putative target site is presented. (b) WT or mutant GCK 3′-UTR luciferase constructs was co-transfected into 293T cells with different doses of hsa-mir-1302 mimic for 40 h. Luciferase assays were performed and luciferase activities were normalized to β-galactosidease activities. Data represent mean±s.d. from three independent experiments. One-way analysis of variance (ANOVA), *P<0.05. (c) WT GCK or mutant GCK plasmid was transfected into 293T cells in the treatment of hsa-mir-1302 inhibitor or scramble for 48 h. Western blotting was performed with anti-GCK. β-Actin expression was used as a control. NS, nonsignificant.
Mutant GCK lowers blood glucose levels at a controlled rate in vivo
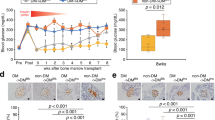
Given our mechanistic understanding behind the activity of the GCK mutant and its expression in vitro, we next applied it in vivo to determine if it was indeed useful at controlling blood glucose levels. We subcutaneously injected GK rats with adenovirus-based WT and mutant GCK and observed survival rate and blood glucose changes for 15 days. It displayed that 40% GK rats treated with WT GCK were dead, compared with no mortality in mutant GCK-treated group (Supplementary Figure 5A). We also found that WT GCK caused more severe drop of blood glucose, whereas mutant GCK made moderate and stable decrease of blood glucose (Supplementary Figure 5B). Later, we investigated if or not adenovirus vector drove the decrease of blood glucose in the STZ-induced diabetic mice. We used adenovirus vector and packaged mutant GCK to infect WT C57BL/6J mice treated with STZ for 8 weeks. The blood glucose levels of mice treated with GCK mutant dropped steadily and significantly throughout the experiment, whereas the mice treated with adenovirus vector had continually elevated blood glucose levels (Figure 4a). Importantly, the mGCK-treated mice did not develop hypoglycemia, with their glucose levels not dropping below that of the negative control.

Mutant GCK moderately decreases blood glucose in vivo. The percentile changes of blood glucose (a) of STZ-induced diabetic mice with or without the treatment of mutant GCK. Data represent mean±s.d. (n=20), unpaired Student’s t-test, *P<0.05 and ***P<0.001. (b) The percentile changes of blood glucose of GK rats with subcutaneous injection of insulin or mutant GCK or GCK vector for 24 h. Data represent mean±s.d. (n=20), one-way analysis of variance (ANOVA), ***P<0.001. (c) GK rats were subcutaneously injected with insulin or mutant GCK or GCK vector at days 0, 1 and 2, and the percentile changes of blood glucose were observed in next 10 days. Data represent mean±s.d. (n=20), one-way ANOVA, ***P<0.001. (d) GK rats were subcutaneously injected with insulin or mutant GCK in 9 days, and the percentile changes of blood glucose were monitored in the next 60 days. Data represent mean±s.d. (n=20), one-way ANOVA, ***P<0.001.
Encouraged by those results, we tested the GCK mutant on GK rats, which normally develop T2D in the absence of obesity,27 and compared its efficacy with that of the conventional insulin treatment. Blood glucose monitoring showed that mutant GCK did not appear to lead to significant changes in the 24 h immediately after subcutaneous injection, whereas insulin therapy led to a sharp decrease (Figure 4b). As such, we next used the GCK mutant in a three-dose series delivered subcutaneously over 3 days, and then tracked the efficacy over 12 days. Reduced blood glucose levels were noticeable by day 4, with that reduction steadily continuing through day 12. In contrast, insulin did not have a prolonged effect, with elevated blood glucose levels returning on day 4 and lasting through day 12. As such, it seems clear that the mutant GCK therapy has the potential for longer-term management (Figure 4c). To further verify that observation, we repeated the experiment with a nine-dose series, and tracked blood glucose levels for 70 days afterwards. Blood glucose levels in the mutant GCK-treated group dipped to normal levels after the doses, and reached a minimum at day 19, before gradually rising through day 70. The slow rise after day 26 was expected given that GK rats naturally increases at 14–15 weeks of age and plateaus after 18 weeks of age, and the rats being used were 10 weeks old at the beginning. Treatment with mutant GCK was more stable and long acting at maintaining lower blood glucose than conventional insulin therapy (Figure 4d).
We also check mGCK level in the liver by analyzing adenovirus particle copies. It showed the highest virus copies in day 1 after ceasing subcutaneous administration of mGCK or vector in 14 consecutive days. Then, there was a decreasing trend revealed in days 7, 14, 21 and 28, which was in accordance with an increasing blood glucose level in the mGCK group and no treatment effect in the mGCK vector group. However, there were no significant differences between the control group and mGCK group at each time point, which ruled out the effects of vector delivery with or with mGCK (Supplementary Figures 6a–c).
Discussion
Diabetes is a chronic condition requiring long-term treatment to control blood glucose levels, with both hyperglycemia and hypoglycemia leading to severe health consequences. As such, methods of treating the condition must be able to be readily maintained over the long term, while causing few large fluctuations. In this paper, we show that the GCK mutant we delivered via gene therapy could have sustained effects over a period of 2 months on GK rats, and the blood glucose level decreased gradually and maintained at a stable level. The vector also did not trigger immune harmful immune effects, a source of concern in all uses of gene therapy. As such, the mutant GCK is likely viable for long-term therapy. Based on our in vitro experiments, we discovered that the molecular mechanisms behind the inhibitory effect are linked to the activities of the microRNA hsa-mir-1302, which may interact with GCK mRNA and impact its stability. We further show that the expression of IL-10 is relevant to the disease severity of diabetes, particularly in terms of glomerulonephritis.
mRNA regulation is being increasingly recognized as being a key element of the regulation of protein expression, and has become the focus of greater attention. One aspect of mRNA regulation that has been of interest is their stability. While generally conceived to be less stable than DNA because of its single-stranded nature, mRNA stability may still vary greatly among different cell types, particularly between 'normal' nucleus-containing cells and anuclear cells such as erythrocytes and platelets. Various factors such as the length of the 3′ cap and 5′ poly-A tail have been previously studied in regard to their ability to control mRNA stability.28, 29 More recently, short-length mirRNAs have been demonstrated to interact with a vast amount of mRNAs, particularly on the untranslated regions on both ends of mRNA (3′- and 5′-UTRs). Such interactions may strongly affect the stability/translation of the mRNA, through direct blockage or indirectly via recruitment of other factors. As several groups have shown however, caution is necessary for positing such mirRNA interactions, and even the existence of the mirRNA of interest itself. After all, not all non-protein coding regions of the genome that contain open reading frames necessarily code for RNA either. With the most obvious examples being the regions of DNA at the telomeres and centromeres, other less evident noncoding regions may also exist. In our present study, we posit a potential mirRNA interaction as being the underlying mechanism for the downregulation of GCK mRNA. We hypothesized such an interaction via sequence analysis, and then supported it through direct in vitro and in vivo experiments. Utilization of microRNA effects to regulate the expression levels of protein may also be applicable for many other gene therapy medications in which such control has proven difficult, by essentially acting as a safer method for adjusting dosage.
There may also be additional immune effects that result from altered expression of GCK that we have been unable to identify conclusively in this paper. After all, many studies have reported that changes in metabolism and metabolic factors can alter the immune response,30 and skew immune balances such as those between M1 and M2 macrophages,31 as well as that between Th17 and regulatory T cells.32 Our results do not show a clear skew against a specific T-helper phenotype, beyond an observation of more marked differences in the STZ model when applied onto IL-10−/− mice. The precise reason behind the increased severity in the absence of IL-10 is not fully clear, but it is likely linked to immune effects that IL-10 may be normally exerting. These effects could include the inhibition of T-effect phenotypes, which would limit their spatial spread and curtail their invasion into the liver. IL-10 may be stimulating regulatory T-cell populations as 'cytokine sinks' or direct inhibitors to advance it. Further work will allow us to more clearly understand the relevant mechanisms.
In our current study, we use gene therapy to deliver a modified GCK to treat T2D. Such a concept of itself is not completely novel, as other groups have attempted to use a similar method to treat diabetes as well. However, direct introduction of the full WT GCK gene was shown to lead to uncontrolled drops in blood glucose levels, likely a result of too much protein being produced. Such an uncontrolled drop would lead to hypoglycemia, a severe and dangerous side effect for diabetic patients. Our novel GCK mutant leads to slower, but more controlled, decreases in blood glucose levels, and did not lead to hypoglycemia in our in vivo murine experiments. We demonstrate a clear rebound in GCK expression levels in STZ-challenged mice to a level lower compared with that induced by infection with the standard WT gene. These results were then further confirmed on GK rats, in which mutant GCK therapy outperformed insulin therapy in the long run, showing that the levels of GCK protein induced by the mutant was sufficient to manage hyperglycemia. The therapy may also be combined with existing therapies, such as insulin, in case of sudden rises of blood glucose levels. Further experiments will be required to confirm its safety, and may also lead to additional improvements in it ability, such as through optimization to more preferentially infect liver and pancreatic cells.
Materials and methods
Mice and rats
C57BL/6J (B6, stock no. 000664), IL10-deficient mice (B6.129P2-Il10tmCgn/J, stock no. 002251), MyD88-deficient mice (B6.129P2(SJL)-Myd88tm.1.1Defr/J, stock no. 009088) and Rag1-deficient mice (B6.129S7-Rag1tm1Mom/J, stock no. 002216) were obtained from Jackson Laboratory (Bar Harbor, ME, USA) and maintained in the barrier facility at the Mount Sinai School of Medicine (New York, NY, USA). For all the experiments, 6–8-week-old female mice were used. The animal study protocols were approved by the Institutional Animal Care and Use Committees of Mount Sinai School of Medicine and Virginia Tech. Male Goto-Kakizaki rats were purchased from SLAC Laboratory Animal Center, Chinese Academy of Science in China. Body weights were around 300±50 g. All of rats experiments were conducted under protocols regulated by the Animal Care Committee of Hangzhou Yiyuan Biotechnology.
Antibodies
The following antibodies were used at the indicated dilutions for immunoblotting and immunoprecipitation. Antibodies for ubiquitin (sc-8017, 1:300) and GCK (sc-7098, WB 1:300, IP 1–2 μg per 100–500 μg of total protein) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-HA-tag antibody (SAB4300603, 1:500) was purchased from Sigma-Aldrich. For flow cytometry, fluorescently labeled antibodies to IL-4 (11B11, fluorescein phycoerythrin labeled), interferon-γ (XMG1.2, allophycocyanin labeled) and CD4 (GK1.5, fluorescein isothiocyanate labeled) were all from eBioscience (San Diego, CA, USA), and were used at a 1:100 dilution. Antibody for IL-17 (TC11-18H10.1, phycoerythrin labeled) was from Biolegend (San Diego, CA, USA), and was used at a 1:100 dilution.
RNA isolation and quantitative real-time RT–PCR
Total RNA was extracted using an RNeasy Plus Kit (Qiagen, Valencia, CA, USA) and cDNA was generated with qScript cDNA Synthesis Kit (Quanta Biosciences, Gaithersburg, MD, USA), followed by analysis using iCycler PCR with SYBR Green PCR Master Mix (Applied Biosystems, Grand Island, NY, USA). Program was chosen to compare the CT value of targeting genes to the housekeeping gene (ubiquitin) in a single sample, using the formula: 10 000x2−ΔΔCT. The following primer sets were used: GCK—sense, 5′-GAAGCGGCCCATGAGGTACT-3′ and antisense, 5′-GTAACAACTCCGCCCCATT-3′; ubiquitin—sense, 5′-TGGCTATTAATTATTCGGTCTGCAT-3′ and antisense, 5′-GCAAGTGGCTAGAGTGCAGAGTAA-3′.
Immunoblotting analysis
Liver homogenate and transfected 293T cells washed with cold phosphate-buffered saline were lysed for 15 min on ice in 0.5 ml of lysis buffer (50 mm Tris-HCl, pH 8.0, 280 mm NaCl, 0.5% Nonidet P-40, 0.2 mm EDTA, 2 mm EGTA, 10% glycerol and 1 mm dithiothreitol) containing protease inhibitors. Cell lysates were clarified by centrifugation (4 °C, 15 min, 14 000 r.p.m.) and the protein was subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting was performed. Antibodies were used according to the manufacturer's instructions. Secondary antibodies were from Santa Cruz Biotechnology.
Transfection and luciferase reporter assay
GCK overexpression plasmids or GCK 3′-UTR luciferase reporter plasmids were transfected into 293T cells in the presence of different treatment. For each transfection, 2.0 μg of plasmid was mixed with 100 μl of Dulbecco's modified Eagle's medium (without serum and antibiotics) and 4.0 μl of Lipofectamine 2000 reagent (Thermo Fisher Scientific, Waltham, MA, USA). The mixture was incubated at room temperature for 20 min and added to 12-well plates containing cells and complete medium. The cells were incubated for 30 h and harvested using reporter lysis buffer (Promega, San Luis Obispo, CA, USA) for determination of luciferase activity. Cells were co-transfected with a β-galactosidase reporter plasmid to normalize experiments for transfection efficiency.
STZ-induced diabetic mice model
Mice received a single intraperitoneal injection of STZ (150 mg kg−1; Sigma-Aldrich, St Louis, MO, USA). Blood glucose and body weight were measured daily. Mice were killed when the body weight dropped to around 20%, and the liver, pancreas, kidney and spleen were harvested for further analysis.
GCK or insulin treatment
Two groups of WT mice (Figure 4a) were induced with single intraperitoneal injection of STZ, and then treated with adenovirus vector (1.65 × 1011 virus particles per kg) or mGCK (1.65 × 1011 virus particles per kg) by six subcutaneous injections every other day. Blood glucose measurement was conducted in the following 52 days.
GK rats were subcutaneously injected with adenovirus vector (1.65 × 1011 virus particles per kg) or insulin (1U kg−1) or mGCK (1.65 × 1011 virus particles per kg) in one dose treatment (Figure 4b) or a three-dose series in 3 days (Figure 4c) or a nine-dose series in 9 days (Figure 4c). Later, blood glucose was measured.
References
Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL et al. A gemome-wide association study of type 2 diabetes in finns detects multiple susceptibility variants. Science 2007; 316: 1341–1345.
Savic D, Ye H, Aneas I, Park SY, Bell GI, Nobrega MA et al. Alterations in TCF7L2 expression define its role as a key regulator of glucose metabolism. Genome Res 2011; 21: 1417–1425.
Stiles B, Wang Y, Stahl A, Bassiliam S, Lee WP, Kim YJ et al. Live-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity. Proc Natl Acad Sci USA 2004; 101: 2082–2087.
Matschinsky FM . Gucokinase as glucose sensor and metabolic signal generator in pancreatic b-cells and hepatocytes. Diabetes 1990; 39: 647–652.
Barzilai N, Rossetti L . Role of glucokinase and glucose-6-phosphatase in the acute and chronic regulation of hepatic glucose fluxes by insulin. J Biol Chem 1993; 268: 25019–25025.
McCarthy MI, Hattersley AT . Molecular diagnostics in monogenic and multifactorial forms of type 2 diabetes. Expert Rev Mol Diagn 2001; 1: 403–412.
McIntyre HD, Ma A, Bird DM, Paterson CA, Ravenscroft PJ, Cameron DP . Metformin increases insulin sensitivity and basal glucose clearance in type 2 (non-insulin dependent) diabetes mellitus. Aust NZ J Med 1991; 21: 714–719.
Proks P, Reimann F, Green N, Gribble F, Ashcroft F . Sulfonylurea simulation of insulin secretion. Diabetes 2002; 51: S368–S376.
Panten U, Burgfeld J, Goerke F, Rennicke M, Schwanstecher M, Wallasch A et al. Control of insulin secretion by sulfonylureas, meglitinide and diazoxide in relation to their binding to the sulfonylurea receptor in pancreastic islets. Biochem Pharmacol 1989; 38: 1217–1229.
Saltiel AR, Olefsky JM . Thiazolidinediones in the treatment of insulin resistance and type II diabetes. Diabetes 1996; 45: 1661–1669.
Katsuno K, Fujimori Y, Takemura Y, Hiratochi M, Itoh F, Komatsu Y et al. Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level. J Pharmacol Exp Ther 2006; 320: 323–330.
Buchwald H, Estok R, Fahrbach K, Banel D, Jensen MD, Pories WJ et al. Weight and type 2 diabetes after bariatric surgery: systematic review and meta analysis. Clin Res study 2009; 122: 248–256.
Buchwald H, Avidor Y, Braunwald E, Jensen MD, Pories W, Fahrbach K et al. Bariatric surgery: systematic review and meta-analysis. J Am Med Assoc 2004; 292: 1724–1737.
Brosky G, Logothetopoulos J . Streptozotocin diabetes in the mouse and guinea pig. Diabetes 1969; 18: 606–611; 5.
Wang Z, Gleichmann H . GLUT2 in pancreatic islets: crucial target molecule in diabetes induced with multiple low doses of streptozotocin in mice. Diabetes 1998; 47: 50–56.
Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J et al. In vitro-expanded antigen specific regulatory T cells suppress autoimmune diabetes. J Exp Med 2004; 199: 1455–1465.
Hammond KJL, Poulton LD, Palmisano LJ, Silveira PA, Godfrey DI, Baxter AG . α/β-T cell receptor (TCR)+ CD4−CD8−(NKT) thymocytes prevent insulin-dependent diabetes mellitus in nonobese diabetic (NOD)/Lt mice by the influence of interleukin (IL)-4 and/or IL-10. J Exp Med 1998; 187: 1047–1056.
Sinuani I, Beberashvili I, Averbukh Z, Sandbank J . Role of IL-10 in the progression of kidney disease. World J Transplant 2013; 3: 91–98.
Kitching AR, Tipping PG, Timoshanko JR, Holdsworth SR . Endogenous interleukin-10 regulates Th1 responses that induce crescentic glomerulonephritis. Kidney Int 2000; 57: 518–525.
Arababadi MK, Mirzaei MR, Sajadi SMA, Hassanshahi G, Ahmadabadi BN, Salehabadi VA et al. Interleukin (IL)-10 gene polymorphisms are associated with type 2 diabetes with and without nephropathy: a study of patients from the southeast region of Iran. Inflammation 2012; 35: 797–802.
Hochstrasser M . Ubiquitin-dependent protein degradation. Annu Rev Genet 1996; 30: 405–439.
Lecker SH, Goldberg AL, Mitch WE . Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 2006; 17: 1807–1819.
Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005; 433: 769–773.
Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E . The role of site accessibility in microRNA target recognition. Nat Genet 2007; 39: 1278–1284.
Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ et al. Combinatorial microRNA target predictions. Nat Genet 2005; 37: 495–500.
Martin HC, Wani S, Steptoe AL, Krishnan K, Nones K, Nourbakhsh E et al. Imperfect centered miRNA binding sites are common and can mediate repression of target mRNAs. Genome Biol 2014; 15: R51.
Wang X, DuBois DC, Cao Y, Jusko WJ, Almon RR et al. Diabetes disease progression in Goto-Kakizaki rats: effects of salsalate treatment. Diabetes Metab Syndr Obes 2014; 7: 381–389.
Mullner EW, Kuhn LC . A stem-loop in the 3’ untranslated region mediates iron-dependent regulation of transferring receptor mRNA stability in the cytoplasm. Cell 1988; 53: 815–825.
Guhaniyogi J, Brewer G . Regulation of mRNA stability in mammalian cells. Gene 2001; 265: 11–23.
Hotamisligil GS . Inflammation and metabolic disorders. Nature 2006; 444: 860–867.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A . Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 2002; 23: 549–555.
Weaver CT, Hatton RD . Interplay between the Th17 and TReg cell lineages: a (co-)evolutionary perspective. Nat Rev Immunol 2009; 9: 883–889.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author contributions
GL and HX initiated the project. HX supervised the project. GL and XT did most of the experiments. ZZ, RZ, LP FZ and JL performed experiments. HH provided critical reagents and gave important advice throughout the study. GL, ZZ and HX wrote the manuscript.
Supplementary Information accompanies this paper on Gene Therapy website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Lu, G., Teng, X., Zheng, Z. et al. Overexpression of a glucokinase point mutant in the treatment of diabetes mellitus. Gene Ther 23, 323–329 (2016). https://doi.org/10.1038/gt.2016.1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2016.1
This article is cited by
-
Citrullination of glucokinase is linked to autoimmune diabetes
Nature Communications (2022)
-
Comprehensive analysis of liver and blood miRNA in precancerous conditions
Scientific Reports (2020)
