
Abstract
Purpose
The recent growth in pan-ethnic expanded carrier screening (ECS) has raised questions about how such panels might be designed and evaluated systematically. Design principles for ECS panels might improve clinical detection of at-risk couples and facilitate objective discussions of panel choice.
Methods
Guided by medical-society statements, we propose a method for the design of ECS panels that aims to maximize the aggregate and per-disease sensitivity and specificity across a range of Mendelian disorders considered serious by a systematic classification scheme. We evaluated this method retrospectively using results from 474,644 de-identified carrier screens. We then constructed several idealized panels to highlight strengths and limitations of different ECS methodologies.
Results
Based on modeled fetal risks for “severe” and “profound” diseases, a commercially available ECS panel (Counsyl) is expected to detect 183 affected conceptuses per 100,000 US births. A screen’s sensitivity is greatly impacted by two factors: (i) the methodology used (e.g., full-exon sequencing finds more affected conceptuses than targeted genotyping) and (ii) the detection rate of the screen for diseases with high prevalence and complex molecular genetics (e.g., fragile X syndrome).
Conclusion
The described approaches enable principled, quantitative evaluation of which diseases and methodologies are appropriate for pan-ethnic expanded carrier screening.
Similar content being viewed by others

Introduction
Carrier screening attempts to identify couples at elevated risk of conceiving a pregnancy affected with a Mendelian condition,1 thereby enabling consideration of alternative reproductive options and early intervention strategies.2, 3, 4 There are thousands of Mendelian conditions1 that differ in both incidence and severity, and for myriad reasons, carrier screening interrogates only a subset of these conditions. Indeed, current guidelines issued by the American Congress of Obstetricians and Gynecologists (ACOG) and the American College of Medical Genetics and Genomics (ACMG)5 suggest pan-ethnic screening only for cystic fibrosis (ACOG and ACMG)6, 7 and spinal muscular atrophy (ACMG).8
Introduced in 2009, “expanded” carrier screening (ECS), which identifies reproductive risks for dozens to hundreds of diseases, has gained acceptance as a reasonable screening approach.5, 9 ECS offerings are diverse, spanning a range of panel sizes and assay technologies. Two ostensibly identical ECS panels with the same number of genes and assay technology may nevertheless differ in their sensitivity due to differences in the number of interrogated positions in each gene and in the interpretation of detected variants. In principle, maximal sensitivity is achieved by determining the sequence at every base in an entire gene and curating all variants to assess pathogenicity. However, due to assay cost and throughput limitations in variant interpretation, the set of interrogated bases is typically limited. In the ECS strategy termed “full-exon sequencing,” most intronic bases are not included in the panel, and next-generation sequencing (NGS) is used to identify all remaining bases across the protein-coding exons and in noncoding regions with known contributions to pathogenesis (e.g., known splice-modifying sites). Full-exon sequencing typically probes thousands of bases per gene and can identify all common variants, plus rare novel variants such as clearly damaging protein-truncating mutations. Because of its potential to discover novel variants, full-exon sequencing requires processes to interpret the clinical impact of all observed variants. An alternative ECS strategy that sidesteps the need for novel-variant curation is “targeted genotyping” (TG), which restricts its focus to a set of predefined pathogenic variants, often between 1 and 50 per gene9 (but in some cases, such as CFTR mutation panels, ranging into hundreds per gene). Although NGS can be used to perform TG, other technologies, such as allele-specific polymerase chain reaction (PCR) and microarray, can also be used. Interestingly, even though TG-based tests may be implemented inexpensively (because of a smaller assayed region and no need for novel variant interpretation), their lower detection rates relative to full-exon sequencing may increase overall health-care spending due to the increased cost of care for undetected affected pregnancies.10
Recently, several professional societies, including ACOG and ACMG, have issued recommendations regarding the broader implementation of ECS. Their suggestions include careful vetting of the clinical and population characteristics of each disease and selection of panel content using “clear criteria, rather than simply including as many diseases as possible.11” Similarly, the European Society for Human Genetics said that “an important screening criterion is that the natural course of the disease screened for should be adequately understood, and that an acceptable and reliable test should be available with known sensitivity, specificity and predictive values.12”
To address these points and increase the transparency of expanded carrier screening panel design, this paper proposes a method for the systematic design of ECS tests. This approach builds on previous work in three main ways: by focusing on diseases with high clinical significance,2, 13 by using well-calibrated curations14 to characterize variants, and by extending a framework suggested previously15 for systematically characterizing the performance of expanded carrier screening panels. We evaluate and discuss this method using ECS data from more than 400,000 patients.
Materials and methods
Systematic design of expanded carrier screening
In this work, we propose the following approach to designing ECS panels:
-
1
Systematically enumerate all candidate diseases for which universal screening is clinically desirable (typically “severe” or “profound” diseases, as defined below).
-
2
Maximize the aggregate panel sensitivity subject to limitations on assay size by selecting candidates that capture a high amount of disease risk (i.e., high-incidence diseases).
-
3
Maximize per-disease sensitivity and negative predictive value (NPV) to yield high confidence in both carrier and noncarrier status for individual conditions.
-
4
Ensure near-100% specificity using carefully designed assay and curation protocols.
Data set summary
Retrospective analysis of anonymized ECS data is exempt from institutional review board (IRB) oversight (as granted by Western IRB on 5 October 2016). All patients provided informed consent for testing and anonymized research. Data are based on de-identified, aggregated ECS results of 474,644 patients who were screened using the Family Prep Screen (Counsyl, South San Francisco, CA) for up to 94 “severe” or “profound” conditions (as described below). For congenital adrenal hyperplasia, we analyzed results only for the classic form and not the nonclassic form, as the nonclassic form is classified as “moderate” severity (as described below). To minimize bias in disease frequencies, patient data were used only if the patient reported no remarkable personal or family history (e.g., infertility, or known history of carrier status or genetic disease). Using a method previously described, data from patients tested under two methodologies (344,743 targeted genotyping-based tests and 129,901 NGS-based tests) were aggregated to reduce statistical uncertainty.15 Statistical calculations were performed using Python 2.7.12, Numpy 1.11.1, and Pandas 0.18.1, as well as additional tools described previously.15 To assess the importance of panel-wide copy-number variant (CNV) deletion calling, data from 56,267 NGS-based tests (primarily a subset of the 129,901 total NGS cohort) were analyzed to estimate the frequencies of exon-or-larger deletions and duplications in 82 genes on the panel (method described in the Supplementary Methods online). Deletions were assumed pathogenic, while duplications were excluded from further analysis. Unless otherwise noted, reported disease risks were weighted by the ethnic distribution of the United States,16 as described in the Supplementary Methods and Supplementary Table S1. Variants of uncertain significance (VUS) are not reported during routine carrier screening and were excluded from the present retrospective analysis (but are included in our assessment of curation in Table 3). Furthermore, variants with known low penetrance or mild phenotype have been excluded from analysis, as described previously (Table S6 in ref. 15). After exclusion of low-penetrance and mild variants, all variants were treated as having equal phenotypic impact (excluding fragile X syndrome, described below). As a case study, we have presented the most prevalent pathogenic variants in GBA in Supplementary Table S5.
Disease selection by clinical severity
An ECS best serves patients by screening for serious diseases.12 Because the classification of large disease lists requires substantial effort on the part of medical professionals, we previously developed2 a rule-based scheme that classifies diseases into increasingly serious categories (“mild,” “moderate,” “severe,” or “profound”), using disease characteristics (e.g., intellectual disability or shortened life span) as inputs. Under this scheme, disease characteristics are grouped into four tiers, with tier 1 being the most serious. This phenotype-based scheme was shown to agree with severity classifications by health-care professionals,2 suggesting its feasibility for efficient classification and comparison of hundreds of diseases. Underscoring the usefulness of such severity classifications, a separate survey of at-risk couples with “severe” or “profound” diseases found that among at-risk couples (wherein both partners are found to be carriers for the same autosomal recessive condition), those at risk for severe or profound conditions altered reproductive decisions at a significantly higher rate than those carrying moderate conditions.13 In the present work, we propose that the first step in ECS disease selection is to enumerate and prioritize diseases considered “severe” or “profound” under this scheme. Although they may not affect reproductive choices, “moderate” conditions (such as GJB2-related hearing loss) may also be candidates for enumeration due to strong clinical desire for testing by patients and providers to enable early preparation. “Mild” conditions are typically screened in an as-requested (rather than routine) fashion. Note that disease characterization is performed at the level of diseases, rather than variants.
Quantifying ECS sensitivity
In our proposed ECS design methodology, diseases are selected in order to maximize the aggregate sensitivity among “severe” and “profound” diseases. Intuitively, this is achieved by selecting diseases with high prevalence; however, the relationship between prevalence and sensitivity must be made precise.
Historically, in the context of ECS panels, disease frequency has often been discussed in terms of carrier frequency and at-risk couple frequency.17, 18 However, these metrics have limitations that complicate their use in assessing sensitivity. Carrier frequency is a suboptimal proxy because a single carrier result alone is not clinically actionable: the reproductive risk is a function of both parents for autosomal recessive diseases. The at-risk couple frequency—frequently the square of carrier frequency for autosomal conditions—is problematic because certain single-gene diseases have complex inheritance patterns that may modulate the risk of transmission to offspring. In fragile X syndrome, for example, fetal risk is not easily derived from the carrier frequency and instead requires a risk model that considers the probability of repeat expansion as a function of maternal CGG repeat number.19 For these reasons, the “modeled fetal disease risk,” defined as the disease probability in a conceptus of randomly selected male and female parents, was recently introduced by Haque and coauthors to quantify the relative yields of different screening panels.15 A complete mathematical and computational definition of disease risk calculation is given in the Supplementary Methods, with disease-specific details described previously (Supporting Information in ref. 15). Modeled fetal disease risk statistically quantifies the rate of affected conceptuses using simulated parental populations and accounts for the various transmission rates of autosomal-recessive, X-linked, and complex (e.g., fragile X syndrome) diseases.
Here we define sensitivity as the fraction of disease risk, summed over all diseases on the panel, that an ECS panel is able to detect:

Here “disease_risk_assessed” is the disease risk captured by a particular panel, while “disease_risk” refers to the “true” disease risk of the population. If a test for a simple autosomal recessive condition were to detect 50% of carriers, the sensitivity would be 50 × 50% = 25%, where the squaring occurs because affected fetuses inherit two damaged copies of a gene. In this case, the test only captures one quarter of the disease risk for that disorder and compromises aggregate sensitivity.
In the present work, the candidate panel disease list is fixed and so the denominator in equation (1) is constant, i.e., it is the population disease risk aggregated over 94 “severe” and “profound” diseases. Therefore, to compare sensitivities of different ECS strategies, we focus on the numerator, i.e., the assessed disease risk.
Clinical accessibility of variants: curation
Genomics assays, particularly those based on NGS, require careful processes for deciding which variants are clinically significant and must be reported to patients. This variant interpretation process directly impacts both sensitivity and specificity. While ACMG and the Association for Molecular Pathology have established general recommendations for this process,14 the optimal interpretation process will depend on the particular disease and subsequent clinical actions being considered.20 In particular, there exists a tradeoff among labor, sensitivity, and specificity in interpretive processes. Although there exist processes involving little labor (e.g., those relying simply on computational methods), it is difficult to achieve both high sensitivity and high specificity simultaneously in a low-labor process.
Supplementary Figure S1 describes the interpretation process developed at Counsyl to serve the needs of a high-throughput population-screening laboratory. Interpretation in this pipeline occurs in “real time,” whereby any novel variant observed in a patient sample (i.e., one that has not otherwise been classified recently) undergoes curation using the above process prior to the release of the patient’s report. The protocol uses an automated pipeline to collect several lines of evidence (population frequency, in silico protein structure predictors, splicing predictors, and conservation) that are then manually reviewed in combination with published case reports and functional studies to determine the variant classification. Prior to manual review, a rule-based system is used to automatically classify variants with high frequency in asymptomatic populations and variants with no literature reports.
To evaluate the variant classification performance of this pipeline, we compared Counsyl Family Prep Screen classifications to ClinVar21 (April 2016 release). As a reference standard, we selected variants that met the following requirements:
-
1
Each variant was classified by Counsyl and at least two of the following nine external clinical labs: ARUP Laboratories, Clinical Biochemistry Laboratory University College London Hospitals, Emory Genetics Laboratory, GeneDx, Genetic Services Laboratory University of Chicago, Invitae, Juha Muilu Group Institute for Molecular Medicine Finland, LabCorp, and Laboratory for Molecular Medicine (Partners HealthCare). These laboratories were chosen due to having public classifications of genes and variants that sufficiently overlap with those on the Counsyl Family Prep Screen.
-
2
Each variant has complete concordance among classifications submitted by external laboratories.
-
3
Cancer susceptibility classifications for ATM and NBN were excluded due to lesser relevance for ECS applications.
The classifications were binned into two simplified categories according to medical management in carrier screening: (i) “pathogenic,” which includes known pathogenic, likely pathogenic, and predicted pathogenic classifications, and (ii) “VUS/benign,” which includes variant of uncertain significance (VUS), known benign, likely benign, and predicted benign. This process identified 505 variants located in 64 genes, of which 179 are pathogenic and 326 are VUS/benign. The molecular impact of the variants was obtained from the Ensembl Variant Effect Predictor (VEP) Web interface for human GRCh37.22
To demonstrate the tradeoff among labor, sensitivity, and specificity, we compared the performance of the widely used VEP algorithm under various sensitivity/specificity thresholds to the performance of a human-in-the-loop interpretive process. Specifically, we compared the Counsyl interpretive pipeline to “VEP-specific,” which prioritizes specificity, by flagging any variant having “high” VEP impact as pathogenic, and “VEP-sensitive,” which prioritizes sensitivity by flagging as pathogenic any variant having “high” or “moderate” impact by VEP.
Results
Compilation and classification of candidate genes
The first step in ECS panel design is the selection and severity classification of a superset of candidate diseases. We performed a preliminary evaluation of 671 diseases and a complete evaluation of 110 diseases—94 “severe” and “profound” conditions used in fetal disease risk calculations and 16 additional conditions.23 As described in Materials and Methods, diseases were classified by severity using objective criteria: as examples, cystic fibrosis and spinal muscular atrophy, recommended for pan-ethnic screening by ACMG are classified as “severe,” whereas GJB2-related DFNB1 nonsyndromic hearing loss and deafness is classified as “moderate.” Severity classifications of select diseases are shown in Table 1, with additional diseases given in Supplementary Table S2.
Disease risk comparison of idealized ECS panels
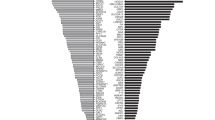
The next steps in our ECS panel-design framework involve maximizing aggregate and gene-level sensitivities. To identify the key factors that influence this maximization, we mined ECS data from 474,644 patients and reweighted the computed risks by the ethnic makeup of the United States.16 The per-gene contribution to the aggregate disease risk assessed by an ECS is shown in Figure 1. It is clear that a few well-known, high-prevalence diseases contribute substantially to the overall disease risk. Furthermore, several of these large contributors, such as FMR1 (fragile X syndrome, 15.8% of total disease risk) and CYP21A2 (CAH; 21-hydroxylase deficient congenital adrenal hyperplasia, 5.2% of total disease risk), arise from genes requiring special care in analysis (Table 2; see Discussion). Over one-fourth (28.9%) of total disease risk is accountable to these four technically challenging conditions, suggesting that in order for an ECS to maximize assessment of disease risk, sensitivity for technically challenging yet highly prevalent diseases is critical.

The disease risk contribution of each severe or profound condition on Counsyl Family Prep Screen is shown for a US census–weighted population. 78 conditions contributing fewer than 2 affected fetuses per 100,000 are lumped into one category (“78 Other”) for visual clarity; individual component diseases are outlined but not labeled. For each condition, the number of affected fetuses (per 100,000) is shown, along with the percentage of the total disease risk. The area of each box is proportional to the disease risk. Reported numbers include panel-wide deletion predictions when applicable (see Supplementary Methods).
To further quantify how inclusion of technically challenging variants impacts aggregate panel sensitivity, we constructed three hypothetical full-exon sequencing ECS panels (Figure 2; top bar) for severe and profound diseases. The baseline panel only calls single-nucleotide polymorphisms, indels, and select high-prevalence CNVs (e.g., founder mutations) and excludes several technically challenging diseases (fragile X syndrome, 21-hydroxylase-deficient congenital adrenal hyperplasia, α-thalassemia, and spinal muscular atrophy; see Discussion and Table 2). The next panel illustrates the marginal gains from adding the technically challenging diseases: excluding these conditions causes 28.9% of affected fetuses to be missed. Finally, the last panel of the top row (Figure 2) assesses the marginal gain of panel-wide exon-level deletion calling (i.e., detection of novel copy-number losses other than known recurrent founder variants). Novel deletion calling adds approximately 4 affected fetuses per 100,000 to the overall detected risk, a relative improvement of 2%. For context, this contribution is roughly comparable to the net contribution of the 50 lowest-prevalence diseases on the panel. The value of novel deletion calling varies by ethnicity: for some ethnicities and diseases, panel-wide deletion calling contributes up to 13 affected fetuses per 100,000 (Supplementary Figures S2 and S3).

The sensitivity of several hypothetical ECS panels is compared using the disease risk as a proxy. The absolute disease risk, in affected fetuses per 100,000, is plotted on the bottom axis. The top axis shows the contribution as a percent of the total assessed disease risk of the most comprehensive panel considered (full-exon sequencing + special cases + CNV). CNV, copy-number variant; ECS, expanded carrier screening.
To quantify the extent to which it is possible to assess disease risk by targeted genotyping (Figure 2; bottom bars), we constructed sensitivity-optimizing hypothetical targeted genotyping assays of different sizes by selecting from a list of variants rank-ordered by their contributions to overall disease risk (see Supplementary Methods). The targeted genotyping assays detect approximately half the maximal disease risk when technically challenging diseases are omitted. A panel consisting of 500 optimally selected variants plus challenging diseases is capable of detecting 92.4% of the total risk. A per-variant analysis of optimal ECS panels is given in Supplementary Figure S4.
To further clarify how sequencing improves risk assessment at the level of individual diseases, we also estimated the per-gene contribution to disease risk for a previous, targeted genotyping version of Family Prep Screen (Supplementary Table S4). Excluding special cases, the targeted genotyping panel detects 95 affected fetuses per 100,000 births; this number lies between the best-case TG panels of 50 and 500 variants, consistent with the panel probing 332 variants. As expected, some of the conditions with a long history of screening are well captured by the TG panel (CFTR: 1.1-fold gain, HBB: 1.08-fold gain), whereas other conditions show larger gains for NGS. In aggregate, using NGS leads to a 1.4-fold increase in disease risk as compared to probing the same diseases with the TG panel. Furthermore, the relative gain of NGS (versus TG) may be greater for ethnicities that are poorly served by existing TG panels, such as cystic fibrosis in Asians (Supplementary Figure S5).
Real-time curation enables full-exon sequencing
While sequencing maximizes analytical sensitivity (relative to genotyping of selected sites), maintaining high clinical sensitivity and specificity also requires accurate variant interpretation. To assess the sensitivity and specificity of real-time ECS variant interpretation, we compared 505 variant classifications using in-house curation, VEP automated curation, and consensus ClinVar submissions (see Materials and Methods). The consensus ClinVar submissions were treated as the reference, although we note that consensus among ClinVar submissions does not necessarily reflect biological truth.
While fully automated computational approaches are much less laborious than full literature-based curation, comparison to ClinVar consensus shows that VEP-based methods must sacrifice either sensitivity or specificity, neither of which is desirable in a screening context (Table 3). In contrast, the integrated interpretation pipeline (see Materials and Methods) achieved both high sensitivity (91.1%) and specificity (99.7%) relative to ClinVar consensus, underscoring the clinical value of combining automated and manual curation.
Discussion
Evaluating expanded carrier screening tests
There is not yet consensus on the “ideal” ECS design. Here, we describe a method in which candidate diseases of high severity are enumerated and then selected for inclusion on a panel based on maximizing aggregate and per-disease sensitivity and specificity. Critically, the optimization of disease-risk sensitivity and specificity differs from a panel-design strategy that simply maximizes the number of genes. Indeed, we argue that disease-risk sensitivity and specificity are better metrics for ECS panel comparisons than gene number, since the latter does not account for features that vary widely across genes, such as disease incidence, assay difficulty, and inheritance pattern.
Assay technology and variant interpretation are key determinants in maximizing ECS sensitivity and specificity. We demonstrate that full-exon sequencing—especially when coupled with technically challenging disease assays and novel CNV detection—provides a sensitivity gain over targeted genotyping. Importantly, these gains require rapid and accurate variant interpretation, which we show can be performed in real time for novel variants in a manner that is consistent across laboratories.
Building an ECS panel requires careful attention to not only what genes to include, but also what genes to exclude. Sequencing, curating, reporting, and counseling on diseases with limited clinical severity may increase health-care costs and heighten patient anxiety without a commensurate improvement in clinical utility. For these reasons, we have advocated the use of a systematic method for assessing disease severity. Although there are limitations and exceptions to such an approach, we argue that it is a useful starting point for comparisons of ECS panels.
While not the main focus of this work, the cost of performing ECS in the clinic remains a key factor when designing an ECS panel. The ECS panel described herein relies almost exclusively on NGS. With a very low cost per base, NGS coupled with appropriate analysis software and an efficient variant interpretation protocol flexibly accommodates panel updates and can affordably assess carrier status even for technically challenging diseases like α thalassemia and CAH, which historically involve more expensive single-gene assays (e.g., multiplex ligation-dependent probe amplification).
The process of ECS panel design should also consider how screening for each disease gene could impact the reproductive options and intervention strategies that patients pursue. For instance, early diet interventions can mitigate the effects of medium-chain acyl-CoA dehydrogenase deficiency and phenylalanine hydroxylase deficiency, making them attractive targets for inclusion in ECS. Similarly, early educational interventions can benefit fragile X syndrome patients,3 underscoring the value of prenatal screening for this condition. Irrespective of whether patients pursue alternative courses of action in response to ECS, the information and autonomy gained from testing holds value.
Technically challenging genes
Several diseases of high clinical importance (Table 2) have low sequence complexity (e.g., CGG repeats) and/or high homology (e.g., pseudogenes),24 making them difficult to assess with targeted genotyping or full-exon sequencing. These typically require either specialized molecular assays (e.g., testing for fragile X syndrome often uses PCR with capillary electrophoresis25), or custom NGS software (e.g., CAH26). As shown in Figures 1 and 2, these “special case” genes contribute substantially to the population disease risk and in turn, ECS test sensitivity. For example, in order to achieve the same sensitivity, a laboratory could either introduce a complex assay for CAH with 95% sensitivity (9.8 affected births per 100,000) or add the 68 rarest conditions (10.0 affected births per 100,000). This comparison highlights an important feature of panel design: panel constitution and panel size both impact assessed disease risk, and the panel with highest assessed disease risk (and therefore clinical value) may not screen the most genes. Therefore, it is important to quantify each gene’s relative impact on the aggregate assessed disease risk of an ECS.
Considerations beyond aggregate sensitivity and specificity
We have suggested using the panel-wide sensitivity and specificity as quantitative comparators of ECS panels. In addition, we suggest that per-disease negative predictive value (NPV) is another critical quantity to consider. As a thought experiment, consider two ECS panels that assess the same aggregate disease risk, say, 100 affected pregnancies per 100,000. Suppose the first panel (panel A) screens 100 diseases with 100% sensitivity, while the second panel (panel B) screens 1,000 genes with 10% sensitivity. Because their aggregate sensitivities are the same, both panels leave couples with the same overall residual risk of an affected pregnancy after a single round of ECS. However, they differ in the disease-level NPV. We argue that panel A is preferable because couples receiving negative results for panel A can be confident that they (i) are not at risk for the diseases listed on the panel and (ii) are at risk only for diseases conspicuously omitted from the panel.
The argument for prioritizing per-disease NPV provides additional support for the inclusion of panel-wide CNV calling and other sensitivity-boosting improvements to a panel’s existing diseases. Recall that the addition of panel-wide large deletion calling increased the captured disease risk by 2%, with each disease receiving a boost in sensitivity. The net sensitivity gain of deletion calling is roughly equivalent to the addition of approximately 50 new genes. If a panel could add CNV calling or 50 additional genes, we argue that the CNV calling is preferable in order to maximize the panel’s disease-level NPV.
Application to future panel design
The present work uses retrospective analysis of a commercial ECS to evaluate recommendations for panel design. These recommendations take the form of both general principles (e.g., maximize the sensitivity, specificity, and per-disease NPV for severe and profound diseases) as well as methodological details (e.g., inclusion of hard-to-sequence genes, use of full-exon sequencing, and calling of novel CNVs).
For future panel design, one will likely not have access to 400,000 test results with clinical-grade variant curations. However, disease incidence data will be available, as well as allele frequency data from, e.g., the Exome Aggregation Consortium.27 These data could be used to drive a similar analysis in a prospective setting. Care will need to be taken due to unknown variant significance, uncertainty in incidence estimates, and unknown assay detection rate. Despite these challenges, we expect the same principles for optimal ECS panel design to hold.
Comparison to ACOG opinion
ACOG very recently released a new opinion31 about carrier-screening panels, making specific recommendations regarding the severity and frequency of diseases that should be screened. Regarding severity, the opinion suggested that screened diseases “have a well-defined phenotype, have a detrimental effect on quality of life, cause cognitive or physical impairment, require surgical or medical intervention, or have an onset early in life.” These criteria are captured well by our disease-severity classification scheme,2 summarized in Table 1. With respect to frequency, the opinion supports screening for diseases with a carrier frequency of 1 in 100 or greater. This carrier frequency threshold roughly corresponds to a disease risk of 2.5 per 100,000 births, which is exceeded by 14 conditions in Figure 1. Although our panel does screen some less frequent conditions, we reiterate that the 78 least prevalent conditions (Figure 1) satisfy the recommended severity criteria and have a combined impact of 17.8 per 100,000 births (10% of total risk), sufficiently in excess of the frequency criteria that we favor their inclusion in ECS. Furthermore, several of the 78 least prevalent conditions do meet the 2.5 per 100,000 threshold when considered on a per-ethnicity basis.
Conclusion
We have described a principled method for ECS panel design that selects candidate diseases using systematic severity classification and maximizes sensitivity among those candidates. Based on laboratory testing data, technically challenging genes and full-exon coverage were identified as dominant contributors to overall sensitivity. Furthermore, we argue that the per-disease negative predictive value is a crucial secondary consideration. Clear principles and methodical panel construction ensure transparency in panel design and address concerns put forth in guidelines from medical organizations. Broader adoption of these or similar methods would result in consistent panel design and establish a clear basis for panel comparison and evaluation by interested health-care providers.
References
Altshuler D, Daly MJ, Lander ES . Genetic mapping in human disease. Science 2008;322:881–888.
Lazarin GA, Hawthorne F, Collins NS et al, Systematic classification of disease severity for evaluation of expanded carrier screening panels. PLoS One 2014;9:e114391.
Hagerman RJ, Berry-Kravis E, Kaufmann WE et al, Advances in the treatment of fragile X syndrome. Pediatrics 2009;123:378–390.
American College of Medical Genetics Newborn Screening Expert Group. Newborn screening: toward a uniform screening panel and system—executive summary. Pediatrics 2006;117:S296–307.
Edwards JG, Feldman G, Goldberg J et al, Expanded carrier screening in reproductive medicine—points to consider: a joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal-Fetal Medicine. Obstet Gynecol 2015;125:653–662.
American College of Obstetricians and Gynecologists Committee on Genetics. ACOG Committee Opinion No. 486: update on carrier screening for cystic fibrosis. Obstet Gynecol 2011;117:1028–1031.
Watson MS, Cutting GR, Desnick RJ et al, Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med 2004;6:387–391.
Professional Practice and Guidelines Committee Prior TW, Professional Practice and Guidelines Committee. Carrier screening for spinal muscular atrophy. Genet Med 2008;10:840–842.
Lazarin GA, Haque IS, Nazareth S et al, An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: results from an ethnically diverse clinical sample of 23,453 individuals. Genet Med 2013;15:178–186.
Azimi M, Schmaus K, Greger V et al, Carrier screening by next-generation sequencing: health benefits and cost effectiveness. Mol Genet Genomic Med 2016;4:292–302.
Grody WW, Thompson BH, Gregg AR et al, ACMG position statement on prenatal/preconception expanded carrier screening. Genet Med 2013;15:482–483.
Henneman L, Borry P, Chokoshvili D et al, Responsible implementation of expanded carrier screening. Eur J Hum Genet 2016;24:e1–e12.
Ghiossi C, Goldberg JD, Haque IS et al, Clinical utility of expanded carrier screening: reproductive behaviors of at-risk couples. bioRxiv 2016. doi:doi:10.1101/069393.
Richards S, Aziz N, Bale S et al, Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424.
Haque IS, Lazarin GA, Kang HP et al, Modeled fetal risk of genetic diseases identified by expanded carrier screening. JAMA 2016;316:734–742.
US Census. http://factfinder.census.gov/faces/nav/jsf/pages/index.xhtml. Accessed 26 January 2016.
Lakeman P, Plass AM, Henneman L et al, Three-month follow-up of Western and non-Western participants in a study on preconceptional ancestry-based carrier couple screening for cystic fibrosis and hemoglobinopathies in the Netherlands. Genet Med 2008;10:820–830.
Professional Practice and Guidelines Committee Gross SJ, Professional Practice and Guidelines Committee Pletcher BA, Professional Practice and Guidelines Committee Monaghan KG, Professional Practice and Guidelines Committee. Carrier screening in individuals of Ashkenazi Jewish descent. Genet Med 2008;10:54–56.
Yrigollen CM, Durbin-Johnson B, Gane L et al, AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet Med 2012;14:729–736.
Adams MC, Evans JP, Henderson GE, Berg JS . The promise and peril of genomic screening in the general population. Genet Med 2016;18:593–599.
Landrum MJ, Lee JM, Benson M et al, ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–D868.
McLaren W, Gil L, Hunt SE et al, The Ensembl Variant Effect Predictor. Genome Biol 2016;17:122.
Nikita Mehta MS, Muzzey D, Leggett M et al, Design of scalable gene panels for carrier screening. ACMG Annual Meeting, 2016. Tampa, FL. http://research.counsyl.com/posters/2016/ACMG/ACMG16-Panel-Design.pdf.
Mandelker D, Schmidt RJ, Ankala A et al, Navigating highly homologous genes in a molecular diagnostic setting: a resource for clinical next-generation sequencing. Genet Med 2016;18:1282–1289.
Chen L, Hadd A, Sah S et al, An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. J Mol Diagn 2010;12:589–600.
Muzzey D . An NGS-based carrier screen for congenital adrenal hyperplasia with 95% detection rate. ASHG Annual Meeting, October 2015, Baltimore, MD.
Lek M, Karczewski K, Minikel E et al, Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291.
Maguire JR, D’Auria KM, Lai HH, et al. Next-generation sequencing carrier screen for Alpha Thalassemia identifies both common and rare variants. AMP Annual Meeting, November 2015. Austin, TX. http://research.counsyl.com/posters/2015/AMP/AMP_AlphaThal.pdf.
D’Auria KM, Theilmann MR, Iori K, et al. NGS-based carrier screen for Gaucher’s disease calls variants and detects large rearrangements between GBA and GBAP1. AMP Annual Meeting, November 2015. Austin, TX. http://research.counsyl.com/posters/2015/AMP/AMP_GBA.pdf.
Wang X, Wan HY, Oyolu CB et al, Next-generation sequencing assay accurately determines carrier status for spinal muscular atrophy ASHG Annual Meeting, October 2015. Baltimore, MD. http://research.counsyl.com/posters/2015/AMP/AMP_SMA.pdf.
American College of Obstetricians and Gynecologists. Carrier screening in the age of genomic medicine. Committee Opinion No. 690. Obstet Gynecol 2017;129:e35–e40.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
All authors except NM, SIC and ISH are employees of Counsyl, a molecular diagnostics laboratory that performs expanded carrier screening. NM, SIC and ISH are former employees of Counsyl. NM is currently an employee of Mayo Clinic. ISH is currently an employee of Freenome.
Additional information
Supplementary material is linked to the online version of the paper at
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Beauchamp, K., Muzzey, D., Wong, K. et al. Systematic design and comparison of expanded carrier screening panels. Genet Med 20, 55–63 (2018). https://doi.org/10.1038/gim.2017.69
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.69
Keywords
This article is cited by
-
Assessment the carrier frequency of monogenic diseases in populations requiring assisted reproductive technology
BMC Medical Genomics (2024)
-
Chinese genetic variation database of inborn errors of metabolism: a systematic review of published variants in 13 genes
Orphanet Journal of Rare Diseases (2023)
-
Populational pan-ethnic screening panel enabled by deep whole genome sequencing
npj Genomic Medicine (2023)
-
Carrier burden of over 300 diseases in Han Chinese identified by expanded carrier testing of 300 couples using assisted reproductive technology
Journal of Assisted Reproduction and Genetics (2023)
-
A capillary electrophoresis-based multiplex PCR assay for expanded carrier screening in the eastern Han Chinese population
npj Genomic Medicine (2022)
