
Abstract
Purpose
Internationally adopted variant interpretation guidelines from the American College of Medical Genetics and Genomics (ACMG) are generic and require disease-specific refinement. Here we developed CardioClassifier (http://www.cardioclassifier.org), a semiautomated decision-support tool for inherited cardiac conditions (ICCs).
Methods
CardioClassifier integrates data retrieved from multiple sources with user-input case-specific information, through an interactive interface, to support variant interpretation. Combining disease- and gene-specific knowledge with variant observations in large cohorts of cases and controls, we refined 14 computational ACMG criteria and created three ICC-specific rules.
Results
We benchmarked CardioClassifier on 57 expertly curated variants and show full retrieval of all computational data, concordantly activating 87.3% of rules. A generic annotation tool identified fewer than half as many clinically actionable variants (64/219 vs. 156/219, Fisher’s P = 1.1 × 10−18), with important false positives, illustrating the critical importance of disease and gene-specific annotations. CardioClassifier identified putatively disease-causing variants in 33.7% of 327 cardiomyopathy cases, comparable with leading ICC laboratories. Through addition of manually curated data, variants found in over 40% of cardiomyopathy cases are fully annotated, without requiring additional user-input data.
Conclusion
CardioClassifier is an ICC-specific decision-support tool that integrates expertly curated computational annotations with case-specific data to generate fast, reproducible, and interactive variant pathogenicity reports, according to best practice guidelines.
Similar content being viewed by others

Introduction
Inherited cardiac conditions (ICCs) represent a major health burden with a combined prevalence of ~1%.1 Genetic testing is recommended to support the management of many ICCs, with roles in diagnosis (particularly valuable for identification of at-risk relatives), prognostication, and therapeutic stratification.
The principal challenge in genetic testing across all diseases is the interpretation of identified sequence variants. This requires evaluation of data from diverse sources, including clinical observations, computational data, and data derived from the literature. Although existing tools aid collection of some of these data types, scientists and clinicians must often access multiple data sources while interpreting a single genetic variant.
The American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) recently released guidelines that aim to standardize variant interpretation.2 These guidelines outline a set of evidence criteria to assess each variant against, along with how these might be weighted and combined to reach a classification. Studies have, however, shown that even when following the ACMG/AMP guidelines, interpretation can differ between different laboratories, with discordance in excess of 10%.3 One key reason for this discordance is the structure of the ACMG/AMP guidelines, which are deliberately broad and lack specific thresholds, to allow adoption across the full spectrum of genetic disorders. As a result, the challenge to individual disease domains is to incorporate expert gene and disease-specific knowledge, to optimize variant interpretation and introduce consensus. Initiatives such as the Clinical Genome Resource (ClinGen)4 are working to define such disease- and gene-specific thresholds, although these are currently limited to pilot phases for specific gene–disease pairs.
The introduction of guidelines, including the logic behind reaching each classification, opens the way for new computational solutions to facilitate their adoption and increase consistency. Indeed, publication of the guidelines has led to the emergence of interactive tools;5,6 however, to date only one of these builds in automation,7 and none incorporate expert disease-specific knowledge.
Here, we describe CardioClassifier, a powerful new tool that utilizes the framework outlined by the ACMG/AMP guidelines, to automatically annotate variants across 17 computational criteria. Each criterion has been individually parametrized for each gene–disease pair using expert disease-specific knowledge. Automated data are integrated with interactively added case-specific information to calculate variant pathogenicity in a fully interactive Web interface that represents a comprehensive variant interpretation platform for ICCs.
Materials and methods
The development and optimization of CardioClassifier is described in three sections:
-
1.
Rule selection and optimization—adapting and parameterizing ACMG/AMP criteria for ICCs
-
2.
Code and implementation
-
3.
Benchmarking CardioClassifier
Rule selection and optimization
For each rule in the ACMG/AMP framework, we first evaluated whether the rule was applicable to the ICC under investigation and, where appropriate, defined more precisely the circumstances under which the rule would be activated. For seven computational criteria (PS1, PM4, PM5, PP3, BA1, BP3, and BP4), parameterization is consistent across all gene–disease pairs. For the remaining criteria, we have incorporated expert disease, gene- and variant type–specific knowledge and data to define thresholds for activation. This includes determination of robust disease-specific maximum frequency thresholds taking into account the genetic architecture of each disease8 (BS1 and PM2; Supplementary Table S1 online), and using large disease cohorts to define both “mutational hotspots”9 (PM1; Figure 1a) and variants observed more frequently in cases when compared with population controls (PS4). As part of this development process, we compared rule activation in CardioClassifier to a set of variants manually curated as part of routine clinical service at the Royal Brompton Hospital (see Supplementary Materials). Full details of how each rule is parameterized can be found in the Supplementary Materials.

(a) Missense variants within a subportion of MYH7, when identified in an HCM patient, have a 97% prior probability of being pathogenic (etiological fraction; EF = 0.97). We activate PM1 for missense variants in this region. Here we use MYH7:c.2221G>T as an example (black bar). (b) Truncating variants in TTN are only known to cause DCM when found in exons constitutively expressed in the heart (proportion spliced in >0.9). We activate PVS1_strong for these variants. Here we use TTN:c.86641delC as an example (black bar). (c) Variants that have been identified as pathogenic in paralogous genes may identify residues that are intolerant to variation. We have created two modified rules, PS1_moderate and PM5_supporting, to incorporate this evidence. Here we use KCNQ1:p.T311I as an example. KCNQ2:p.T276I is associated with Ohtahara syndrome. We activate PS1_moderate for KCNQ1:p.T311I, which is the equivalent missense change (i.e., same reference and alternate amino acids) in a different member of the same protein family.
As most large reference populations, such as ExAC,10 are not comprehensively screened for health, disease-associated alleles may be observed at low frequency. This holds true for ICCs, which can be difficult to detect even with targeted investigation, as they often manifest later in life and exhibit incomplete penetrance. We have therefore modified PM2 so as not to inappropriately discard variants seen at very low frequencies in these reference data sets.
In addition, we have created extensions to three ACMG/AMP rules, to enhance interpretation of ICC variants. First, we have modified PVS1 for the titin (TTN) gene, which has a role in up to 20% of dilated cardiomyopathy (DCM) cases.11 We have previously shown that only TTN truncating variants in exons constitutively expressed in the heart are robustly associated with DCM.10 Additionally, it is unclear that the mechanism of action for these variants is truly loss of function. Instead of scoring all TTN truncating variants equally and assuming an underlying loss-of-function mechanism, we only score TTN truncating variants highly if they are in constitutive exons (proportion spliced in >0.9; Figure 1b), and we reduce the strength of evidence by one level from very strong to strong (coded as PVS1_strong).
We also extended PS1 and PM5 to utilize known disease-causing variants in related genes/proteins (paralogues) to identify residues intolerant to variation12 (Figure 1c). Where nothing is known about variants at the equivalent residue of the same gene, we use high confidence variants (i.e., same reference allele and M-coffee mapping score >3) as evidence if they affect the equivalent residue in a paralogue (with the same reference allele), either with the same substitution (rule PS1_moderate—Equivalent amino acid change as an established pathogenic variant in a paralogous gene), or a different substitution (rule PM5_supporting—Missense change at an amino acid residue where a pathogenic missense change has been seen in the equivalent residue of a paralogous gene). This analysis is currently restricted to the families of predominantly ion channel proteins associated with inherited arrhythmia syndromes for which this method has been previously validated.12,13
We have previously shown paralogue annotation to be informative for over one-third of novel single-nucleotide variants,13 and independent validation has shown a high specificity and positive predictive value compared with other sources of evidence.14,15 To determine the effect of these criteria on variant classification (before inclusion of any case-level or functional data that cannot be computationally predicted) we used 48 clinically curated (i.e., not literature only or research) missense variants from ClinVar identified as pathogenic or likely pathogenic for long QT syndrome from one or more submitter with at least one review status star, and compared CardioClassifier interpretations with and without paralogue data. Paralogue data were available for 11/48 (22.9%) variants and resulted in a potential change of class from variant of uncertain significance (VUS) to likely pathogenic for 63.6% (7/11) of these (Supplementary Table S2).
Code and implementation
CardioClassifier is implemented server-side in perl and PHP. Uploaded variant data is annotated by the Ensembl Variant Effect Predictor16 and converted to a table using the tableize_vcf.py script within LOFTEE (https://github.com/konradjk/loftee). Protein-altering and splice-site variants (coding ±8 bps) are analyzed for a set of 40 genes associated with inherited cardiac conditions (Table 1). We look to continuously expand this list, focusing on curated genes robustly implicated in disease, emerging from community efforts such as ClinGen.4
The classifier automatically assesses each variant for 17 rules across three distinct data categories, as defined by the ACMG/AMP guidelines.2 It also consults an internal knowledge base of additional evidence, grouped by ACMG rule, either derived from community curation efforts or manually curated internally. The output is displayed on a PHP Web page that allows the user to interact and add (or remove) additional levels of evidence.
Benchmarking
Data sets
To test CardioClassifier extensively we used data from the following sources:
-
1.
ClinVar—all variants identified as pathogenic or likely pathogenic by multiple submitters with no conflicting data (i.e., no reports of benign, likely benign, or uncertain significance) for hypertrophic cardiomyopathy (HCM; n = 158), dilated cardiomyopathy (DCM; n = 16), long QT syndrome (n = 18), catecholaminergic polymorphic ventricular tachycardia (n = 1), Brugada syndrome (n = 4), or arrhythmogenic right ventricular cardiomyopathy (n = 22) were extracted from the 20161201 release of ClinVar17 using publicly available scripts.18
-
2.
Fifty-seven protein-altering variants in MYH7 that have been expertly curated by the ClinGen Inherited Cardiomyopathy expert panel (https://www.ncbi.nlm.nih.gov/clinvar/submitters/506161/).
-
3.
A prospective data set of 327 HCM cases and 625 healthy volunteers recruited to the National Institute for Health Research Royal Brompton cardiovascular biomedical research unit, all phenotypically characterized using cardiac magnetic resonance imaging. Samples were sequenced using the Illumina (San Diego, USA) TruSight Cardio Sequencing Kit1 on the Illumina (San Diego, USA) NextSeq platform. This study had ethical approval (REC: 09/H0504/104+5) and informed consent was obtained for all subjects.
Comparison with existing resources
We compared the performance of CardioClassifier against the generic tool InterVar,7 to assess the importance of our disease-specific annotations. We used the ClinVar data set of 219 variants described above as a test data set.
InterVar scripts were downloaded from GitHub (https://github.com/WGLab/InterVar) and individually run for each disease using an engineered VCF file. To ensure a fair comparison, we edited the “disorder_cutoff” to be equivalent to the thresholds used to activate BS1 in CardioClassifier. All other settings were left as default and no additional evidence was uploaded. We compared both the final classifications and the individual rules that were activated by each tool.
Code and tool availability
CardioClassifier is available at www.cardioclassifier.org, with a free license for noncommercial use. The code and data used to produce this manuscript are available at https://github.com/ImperialCardioGenetics/CardioClassifierManuscript.
Results
Semiautomation leads to high-quality and reproducible variant interpretation
CardioClassifier provides a simple-to-use Web interface that takes as input either individual variant details or a single sample VCF (Supplementary Figure S1). Users select one of 11 cardiac disorders, and this determines which prespecified validated disease genes are analyzed. Where a diagnosis is uncertain (e.g., sudden cardiac death or complex cardiomyopathy), a wider analysis can be performed for genes associated with a broader phenotype (e.g., all cardiomyopathies, or all arrhythmia syndromes; Table 1), or for all 40 ICC genes parameterized. Details of the key features of CardioClassifier can be found in Table 2.
Each variant is annotated for up to 17 computational criteria, with results output to a grid representing the ACMG/AMP framework (Figure 2). The variant report is interactive, allowing a user to add additional case-level evidence to generate and refine a final classification (Supplementary Figure S2). The report is transparent, with all supporting evidence displayed along with links out to eight external resources that are commonly used for interpretation of ICC variants: the ExAC browser,19 Ensembl, the University of California–Santa Cruz Genome Browser, ClinVar, PubMed, Google, the Beacon Network (https://beacon-network.org), and the Atlas of Cardiac Genetic Variation.9

A grid is output for each individual variant. Rules highlighted in color are activated for the variant and rules in gray on a white background are assessed but not activated. A user can click on a rule to manually add or remove a piece of evidence. All evidence used to assess the variant is displayed under the grid along with links out to external resources. An overall classification for the variant using the American College of Medical Genetics and Genomics/Association for Molecular Pathology logic is displayed in the top left corner. EF, etiological fraction (the prior probability that a variant, identified in a case, is pathogenic).9 CADD, combined annotation dependent depletion; HCM, hypertrophic cardiomyopathy; HGVS, Human Genome Variation Society; LOF, loss of function; MAF, minor allele frequency; CGG, Imperial Cardiovascular Genetics and Genomics; ESP, NHLBI Exome Sequencing Project; FH, Family history; PSI, Proportion spliced in; FATHMM, Functional analysis through Hidden Markov Models..
Highly curated data sets of disease cases and healthy controls aid annotation and filtering
As well as publicly available data for both cases and population controls, CardioClassifier incorporates data from three highly curated in-house data sets sequenced with the Illumina TruSight Cardio sequencing panel.1 Counts from 877 DCM, 327 HCM cases, and 1,383 healthy volunteers, all rigorously phenotyped using cardiac magnetic resonance imaging, are used to annotate variants in genes associated with these disorders.
Some genomic regions, especially those that are repetitive or with high GC content, are not fully covered by standard exome sequencing used by major reference data sets. Specifically, 12.5% of sample bases across our 40 ICC genes are covered at <20 × (Supplementary Figure S3) in the ExAC data set. In contrast, our control set has 99.9% of sample bases covered at >20 ×, allowing accurate identification of common and low-frequency variants and platform-specific errors, across all regions of interest (rule BS1). As this data set is derived from the Illumina TruSight Cardio sequencing panel, users uploading variants derived from different sequencing panels should consider comparison with a local data set to identify platform-specific errors.
In addition to these in-house data, we display counts from published clinical cohorts for HCM,9,20 DCM,9,21 long QT syndrome,22 and Brugada syndrome.23 These data are also used to assess individual variants for enrichment in cases over controls (rule PS4).
Results show high concordance with manually curated and gold-standard data
We compared CardioClassifier with 57 gold-standard, manually curated protein-altering variants in MYH7 that have been expertly curated by the ClinGen Inherited Cardiomyopathy Expert Panel.24 Of 222 rules activated by ClinGen for these 57 variants, 157 represented computationally accessible data (from 9 ACMG/AMP rules) that were fully retrieved by CardioClassifier. CardioClassifier concordantly activated 137/157 rules (87.3%; Figure 3a; Supplementary Table S3). The discrepancies fall across 3 rules: PP3 (in silico prediction algorithms; n = 12), PS4 (prevalence in affected individuals statistically increased over controls; n = 7), and PM5 (same amino acid residue as known pathogenic variant; n = 1). CardioClassifier imposes a more stringent threshold on PP3 (allowing only one of eight in silico prediction algorithms to be discordant), and differences in PS4 and PM5 are due to the increased availability of proband data to the ClinGen team (not available from public repositories). In all cases, CardioClassifier successfully returned all available data.

(a) Comparing CardioClassifier to a set of 57 MYH7 expert panel curated variants. Rules were split into those that can be computationally annotated and those that are “case-level” and require manual input. CardioClassifier was run using an “All Cardiomyopathy” test to reflect the spectrum of phenotypes caused by variants in MYH7. *Of the computational rules, 3 were removed from the comparison as they represent draft modifications to the American College of Medical Genetics and Genomics framework by the ClinGen Cardiovascular Domain Working Group that were not published at the time of this work, and not yet implemented in CardioClassifier. Specifically, truncating variants in MYH7 activate a new rule, PVS1_moderate. Additionally, for variants classified as benign by frequency alone (BA1) CardioClassifier does not assess any further rules, leading us to remove an additional data point from the comparison as we would not expect it to be retrieved. (b) Counts of individual rules activated by CardioClassifier and InterVar for 219 variants identified as pathogenic or likely pathogenic in ClinVar. Only pathogenic evidence rules and rules activated by one of the tools at least once are shown.
We then tested the ability of the links within the CardioClassifier report to inform activation of the 61 case-level data points activated by the ClinGen team. These links allowed us to manually collate 50/61 (82.0%) individual data points (Supplementary Table S3) with differences again in the availability of proband data (6 PS4_supporting, 1 PS4_moderate, 1 PS2, 1 BS4, and 2 PP1_moderate). After addition of this clinical data, we reached an identical classification to the ClinGen team for 50/57 (87.7%) variants (Figure 3a).
CardioClassifier has higher sensitivity and specificity than nonspecific interpretation tools
In February 2017 InterVar, and its companion Web server winterVar, became the first tools to automatically populate criteria from the ACMG/AMP guidelines.7 While these tools were crucial steps forward in application of the framework, they aim to support interpretation across the full spectrum of human genes and disorders.
To determine the added value of the disease- and gene-specific annotations included in CardioClassifier, we compared CardioClassifier with InterVar using a set of 219 variants identified as pathogenic or likely pathogenic on ClinVar, with high confidence, across six ICCs. Based on automatically retrieved data only, InterVar identified 64/219 (29.2%) variants as likely pathogenic or pathogenic, while CardioClassifier identified over double this number as clinically actionable (156/219) with a sensitivity of 71.2% (Supplementary Table S4). For both tools, sensitivity would be increased further through user addition of clinical and functional data.
Despite the lower sensitivity of InterVar, there are occasions where the tool activates rules inappropriately in the absence of gene-specific knowledge. First, InterVar activates PVS1 in the TTN gene, regardless of protein location, when it is recognized that truncating variants in exons not constitutively expressed in the heart are not associated with DCM, and are commonly found in demonstrably healthy controls.11 Consequently, InterVar will categorize rare variants in these regions as likely pathogenic when they are highly unlikely to be disease-causing.
Second, InterVar activates rule PP5 (reputable source identifies the variant as pathogenic) for 89.5% of the variants as they are reported as pathogenic in ClinVar. The ACMG guidelines state that this rule should only be activated when the evidence supporting the classification is unavailable, yet this evidence is often contained within the appropriate ClinVar submission. Full details of the rules activated by both tools are shown in Figure 3b.
To ensure the higher sensitivity of CardioClassifier was not due to overactivating rules, we also tested a set of 67 benign and likely benign variants from ClinVar across the same six ICCs. CardioClassifier identified 61/67 (91.0%) of these as benign and the remaining 6 as VUS. Conversely, InterVar identified 41/67 (61.2%) as benign with 22 as likely benign and 4 as VUS. Here InterVar activates BS2 when a variant is seen in the 1000 Genomes data set, which we believe is inappropriate for ICCs that do not fit the important caveat of “full penetrance expected at an early age.” We do acknowledge, however, that InterVar was developed for severe congenital and very early–onset developmental disorders with nearly 100% penetrance.
Diagnostic yield in HCM cases matches previous reports
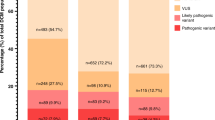
To investigate the clinical utility of CardioClassifier we used a data set of 327 HCM cases. In 66 cases (20.2%) we identified a pathogenic (n = 11) or likely pathogenic (n = 55) variant, with a further 76 cases (23.2%) harboring a VUS. To determine the proportion of these VUS likely to become clinically actionable after the addition of case-level data, we calculated the excess of VUS in cases over the background level of rare and presumably benign VUS in 625 healthy volunteers. Based on a background level of 9.7%, we calculate a case excess of VUS of 13.5%. Combining this with the 20.2% of cases with a pathogenic or likely pathogenic variant, overall, 33.7% of cases have a potentially clinically relevant variant (Supplementary Figure S4a), comparable with previous reports.20
Manual curation of known variants
In addition to automatic retrieval of computational data, CardioClassifier will store curated case-level data entered by users, or prepopulated by active curation. We have primed this “knowledge base” with data from 120 fully curated cardiomyopathy variants, comprising the 57 expert panel curated MYH7 variants and the most commonly observed variants for the major cardiomyopathies: HCM, DCM, and arrhythmogenic right ventricular cardiomyopathy, defined as those occurring six or more times in the Atlas of Cardiac Genetic Variation resource (reflecting a HCM case frequency of ~1/1,000).9 There were 84 such recurrent variants in the Atlas, together representing 39.5% (1,258/3,186) of all identified variants. We curated 63 that had not already been assessed by the expert panel.
After manual curation of the literature and ClinVar for reports of segregation, de novo occurrence, and functional characterization, 34 variants were classified as pathogenic, 13 as likely pathogenic, and 7 as VUS (Supplementary Table S5; Supplementary Figure S4b). The annotations for these 120 variants, accounting for at least 40% of variants identified in Caucasian cardiomyopathy cases, are stored in CardioClassifier, ensuring these variants are correctly classified without further user input.
Discussion
We describe CardioClassifier, an automated and interactive Web tool to aid clinical variant interpretation across a wide range of ICCs. To the best of our knowledge this represents a unique disease-specific solution that automates data retrieval, incorporates gene- and disease-specific knowledge to refine rule application, is preloaded with curated data on prior observations (in health or disease), and integrates evidence according to the widely adopted framework from ACMG/AMP. The tool is transparent, with all the information incorporated into interpreting each variant displayed along with the final classification. It is also flexible, and designed to be fully interactive, with the user able to add and remove evidence specific to the patient/family of interest.
The strength of CardioClassifier is its disease specificity. The ACMG/AMP rules are intentionally nonspecific to allow adoption in any disease domain. To harness the full power of this framework, the rules need to be applied in a disease- and gene-specific manner.25 We have defined criteria and thresholds for each ACMG/AMP rule that are specific to the disorder of interest, and demonstrate the power and effectiveness of this approach over a recently released genome-wide interpretation interface. Incorporation of disease-specific knowledge is limited by current data, and the power of this tool will increase over time as new data become available.
Ongoing community initiatives, such as the Clinical Genome Resource (ClinGen), are defining consensus disease- and gene-specific standards for modifications to the ACMG/AMP guidelines, and it is our intention to continue to develop CardioClassifier to utilize these standards as they become available.
We believe the main limitation to the effectiveness of any computational solution is the retrieval of clinical and patient-specific data that is seldom available as fully structured data for programmatic retrieval. CardioClassifier combines prepopulated computational data with interactive addition of case- and variant-specific evidence in a structured format to overcome this hurdle. Our growing variant knowledge base will add to available structured representation of this crucial case-level data. Future development of CardioClassifier will streamline data-sharing, expanding our knowledge base and sharing it with the community via submission to the ClinVar database. This increasing knowledge base relies on researchers and clinicians in the field supporting data-sharing initiatives, and facilitating direct ClinVar submission from CardioClassifier for the benefit of the ICC community is a development priority.
A further limitation to CardioClassifier in its current form is the restricted prediction of impact on splicing. This arises for two main reasons. First, CardioClassifier uses the Ensembl Variant Effect Predictor to annotate variants, which annotates bases within 8 base-pairs of the exon/intron boundary as splice site, but will miss more distal bona fide splice-site variants. Second, we currently have not incorporated any in silico splice-site prediction algorithms, due to limitations around availability, licensing, and accuracy. These issues will be addressed in a future release.
CardioClassifier is designed to work seamlessly with data from any sequencing platform in standard VCF format, whether targeted sequencing (e.g., Illumina TruSight Cardio1), or targeted analysis of exome-/genome-wide data. This is a crucial step in broadening the availability of genetic testing for ICCs, and standardizing variant interpretation in this field. Furthermore, we hope that in demonstrating the clinical utility of our disease-specific approach, we will encourage others to develop similar tools across other disease specialties.
References
Pua CJ, Bhalshankar J, Miao K et al. Development of a comprehensive sequencing assay for inherited cardiac condition genes. J Cardiovasc Transl Res 2016;9:3–11.
Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–423.
Harrison SM, Dolinsky JS, Johnson AEK et al. Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet Med 2017;19:1096–1104.
Rehm HL, Berg JS, Brooks LD et al. ClinGen the clinical genome resource. N Engl J Med 2015;372:2235–2242.
Patel RY, Shah N, Jackson AR et al. ClinGen pathogenicity calculator: a configurable system for assessing pathogenicity of genetic variants. Genome Med 2017;9::3.
Kleinberger J, Maloney KA, Pollin TI, Jeng LJB. An openly available online tool for implementing the ACMG/AMP standards and guidelines for the interpretation of sequence variants. Genet Med 2016;18:1165–1165.
Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet 2017;100:267–280.
Whiffin N, Minikel E, Walsh R et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med 2017;19:1151–1158.
Walsh R, Thomson KL, Ware JS et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med 2016;19:192–203.
Lek M, Karczewski KJ, Minikel EV et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291.
Roberts AM, Ware JS, Herman DS et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med 2015;7:270ra6–270ra6.
Ware JS, Walsh R, Cunningham F, Birney E, Cook SA. Paralogous annotation of disease-causing variants in long QT syndrome genes. Hum Mutat 2012;33:1188–1191.
Walsh R, Peters NS, Cook SA, Ware JS. Paralogue annotation identifies novel pathogenic variants in patients with Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia. J Med Genet 2013;51:35–44.
Kapplinger JD, Tseng AS, Salisbury BA et al. Enhancing the predictive power of mutations in the C-terminus of the KCNQ1-encoded kv7.1 voltage-gated potassium channel. J Cardiovasc Transl Res 2015;8:187–197.
Kapplinger JD, Giudicessi JR, Ye D et al. Enhanced classification of Brugada syndrome-associated and long-QT syndrome-associated genetic variants in the SCN5A-encoded nav1.5 cardiac sodium channel. Circ Cardiovasc Genet 2015;8:582–595.
McLaren W, Gil L, Hunt SE et al. The Ensembl Variant Effect Predictor. Genome Biol 2016;17:122.
Landrum MJ, Lee JM, Riley GR et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2013;42:D980–D985.
Zhang X, Minikel EV, O-Luria AH, MacArthur DG, Ware JS, Weisburd B. ClinVar data parsing. Wellcome Open Res 2017;2:33.
Karczewski KJ, Weisburd B, Thomas B et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res 2016;45:D840–D845.
Alfares AA, Kelly MA, McDermott G et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med 2015;17:880–888.
Pugh TJ, Kelly MA, Gowrisankar S et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med 2014;16:601–608.
Kapplinger JD, Tester DJ, Salisbury BA et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6:1297–1303.
Kapplinger JD, Tester DJ, Alders M et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010;7:33–46.
Kelly MA, Caleshu C, Morales A et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inheritedcardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet Med 2017; Jan 4 [Epub ahead of print].
Amendola LM, Jarvik GP, Leo MC et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet 2016;99:247.
Acknowledgments
This work was supported by the Wellcome Trust (107469/Z/15/Z), the Medical Research Council (United Kingdom), the National Institute for Health Research (NIHR) Cardiovascular Biomedical Research Unit at Royal Brompton and Harefield NHS Foundation Trust and Imperial College London, the Royal Brompton & Harefield Cardiovascular Research Centre Biobank, the NIHR Biomedical Research Centre at Imperial College Healthcare NHS Trust and Imperial College London, Fondation Leducq (11 CVD-01), the British Heart Foundation (SP/10/10/28431, FS/15/81/31817), and a Health Innovation Challenge Fund award from the Wellcome Trust and Department of Health (United Kingdom) (HICF-R6-373).
This publication includes independent research commissioned by the Health Innovation Challenge Fund, a parallel funding partnership between the Department of Health and the Wellcome Trust. The views expressed in this work are those of the authors and not necessarily those of the Department of Health or the Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflict of interest.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Whiffin, N., Walsh, R., Govind, R. et al. CardioClassifier: disease- and gene-specific computational decision support for clinical genome interpretation. Genet Med 20, 1246–1254 (2018). https://doi.org/10.1038/gim.2017.258
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.258
Keywords
This article is cited by
-
Investigation of mutation spectrum amongst patients with familial primary cardiomyopathy using targeted NGS in Indian population
Journal of Applied Genetics (2024)
-
An AI-based approach driven by genotypes and phenotypes to uplift the diagnostic yield of genetic diseases
Human Genetics (2024)
-
A machine learning approach based on ACMG/AMP guidelines for genomic variant classification and prioritization
Scientific Reports (2022)
-
Analysis of clinically relevant variants from ancestrally diverse Asian genomes
Nature Communications (2022)
-
Genome-wide association analyses identify new Brugada syndrome risk loci and highlight a new mechanism of sodium channel regulation in disease susceptibility
Nature Genetics (2022)
