
Abstract
Purpose
We aimed to identify the cause of disease in patients suffering from a distinctive, atypical form of Usher syndrome.
Methods
Whole-exome and genome sequencing were performed in five patients from three families of Yemenite Jewish origin, suffering from distinctive retinal degeneration phenotype and sensorineural hearing loss. Functional analysis of the wild-type and mutant proteins was performed in human fibrosarcoma cells.
Results
We identified a homozygous founder missense variant, c.133G>T (p.D45Y) in arylsulfatase G (ARSG). All patients shared a distinctive retinal phenotype with ring-shaped atrophy along the arcades engirdling the fovea, resulting in ring scotoma. In addition, patients developed moderate to severe sensorineural hearing loss. Both vision and hearing loss appeared around the age of 40 years. The identified variant affected a fully conserved amino acid that is part of the catalytic site of the enzyme. Functional analysis of the wild-type and mutant proteins showed no basal activity of p.D45Y.
Conclusion
Homozygosity for ARSG-p.D45Y in humans leads to protein dysfunction, causing an atypical combination of late-onset Usher syndrome. Although there is no evidence for generalized clinical manifestations of lysosomal storage diseases in this set of patients, we cannot rule out the possibility that mild and late-onset symptoms may appear.
Similar content being viewed by others

Introduction
Usher syndrome (USH) is the leading cause of inherited deaf-blindness (MIM 268000). It is an autosomal recessive disease with symptoms including visual disturbance due to retinal degeneration and sensorineural hearing loss (SNHL) accompanied in some cases by vestibular areflexia. The prevalence of USH has been estimated as 3.5–16.6 in 100,000 in several populations.1,2,3 USH has historically been classified into three subtypes according to the age of onset of the visual and auditory symptoms, the severity of hearing loss, and the presence of vestibular involvement: Usher type 1 (USH1) is characterized by congenital SNHL with vestibular areflexia and retinitis pigmentosa (RP) onset in the first decade of life; USH2 by mild to moderate nonprogressive SNHL with RP onset in the first or second decade of life and normal vestibular function; and USH3 by progressive SNHL, sometimes accompanied by vestibular involvement, with a variable time of onset of both SNHL and RP. However, cumulative data have identified a large number of families with phenotypes that are not compatible with any of the three established types and are therefore considered as having atypical forms of USH.4,5,6
USH is highly heterogeneous, not only clinically but also genetically. Thirteen causative genes have been identified to date, and three more loci are suspected to include additional genes (RETNET database at https://sph.uth.edu/RetNet/). The development of high-throughput sequencing techniques, including whole-exome sequencing (WES) and whole-genome sequencing (WGS), has contributed tremendously to the identification of novel causative mutations and genes for rare inherited diseases in general and retinal degeneration in particular.7,8 Nevertheless, despite these advances, the causative gene remains unknown in a large number of cases with inherited retinal disease (IRD).9,10 Some of the proteins encoded by USH-associated genes interact in complexes that are localized to hair cells of the inner ear and in retinal photoreceptors.11,12,13 USH-related proteins perform a variety of cellular functions, including myosin-based motor transport (MYO7A); cell–cell adhesion (CDH23 and PCDH15), scaffold proteins (USH1C, ADGRV1, and SANS), and ciliary function (ADGRV1 and C-Nap1).4,12,14 In the present study, we used a combination of WES and WGS in a set of patients who share the same ethnicity and the same distinctive phenotype in order to identify the molecular cause of atypical USH.
Materials and methods
Patient recruitment
Three Yemenite Jewish families including five individuals with USH (two patients in family MOL0120, two in family MOL0737, and one in family TB55) were recruited for this study. The tenets of the Declaration of Helsinki were followed, the study was approved by the institutional review boards of our respective institutions, and before donation of a blood sample, written informed consent was obtained from every individual who participated in this study.
Ocular examination
Ocular evaluation included a comprehensive ophthalmologic exam, Goldmann perimetry, full-field electroretinography (ffERG), electrooculography, color vision testing using the Ishihara 38-panel and Farnsworth-Munsell D-15 tests and color and pseudocolor fundus photos, optical coherence tomography, and fundus autofluorescence (FAF) imaging.
Electrophysiological testing
International Society for Clinical Electrophysiology of Vision standard ffERG was performed using monopolar corneal electrodes (Henkes type; Medical Workshop, Groningen, The Netherlands) and a computerized system (UTAS 3000; LKC, Gaithersburg, MD). Cone responses to 30-Hz flashes of white light were acquired under a background light of 21 cd/m2. Scotopic responses, including a rod response to a dim blue flash and a mixed cone–rod response to a standard white flash, were acquired after 30–45 min of dark adaptation. Between two and four sets of responses were recorded in each condition for reproducibility. All ffERG responses were filtered at 0.3 to 500 Hz, and signal averaging was applied.
Audiometry testing
Audiological screening included a comprehensive questionnaire (collecting information regarding any history of exposure to noise, ototoxic agents, and genetic factors related to hearing impairment), as well as physical and audiometric examinations. An age-appropriate audiological examination was performed, including pure-tone audiometry (250 Hz to 12,000 Hz), tympanometry, and transient-evoked otoacoustic emission (TEOAE) for each ear. We used the following scale to grade the degree of SNHL: slight, 16–25 dB hearing loss; mild, 26–40 dB; moderate, 41–55 dB; moderately severe, 56–70 dB; severe, 71–90 dB, and profound, over 90 dB. Specific types of SNHL were determined by audiometric curve patterns: ascending (hearing loss greater at the lower frequencies), flat, descending (hearing loss greater at the higher frequencies), and U-shaped (hearing loss at mid-frequencies) curves. Otoacoustic emissions have been suggested as a sensitive measure of cochlear function with the potential for preclinical detection of damage. TEOAE tests were conducted on the same day as the pure tone test. A response at three frequencies of 3 dB or greater above the background with a minimum of 70% reproducibility at each frequency and 90% or greater stability was required for passing the TEOAE test. TEOAE examination was categorized as either pass or fail for each ear.
Genetic analyses
Homozygosity mapping was performed using whole-genome single-nucleotide polymorphism microarrays (Affymetrix 6.0, USA and Illumina, San Diego, CA) and analyzed using HomozygosityMapper (http://www.homozygositymapper.org). WES analysis of individuals MOL0120 III:2 and MOL0737 II:2 was performed at Otogenetics (Norcross, GA) using Agilent V4 (Santa Clara, CA, USA) and Illumina HiSeq 2000 with 30 × coverage. WES analysis of individual TB55 II:1 was performed at the Technion Genome Center, using the Nextera Expanded Exome Enrichment Kit (Illumina, USA) and Illumina HiSeq2500 at 30 × coverage. Sequence reads were aligned to the human genome reference sequence (hg38), and variant filtering analysis was performed.
WGS
WGS was performed using the Complete Genomics platform (format version 2.5), and the data were analyzed as described previously.9
Primer design
Primers for mutation screening were designed using the Primer3 software and Sanger sequencing of polymerase chain reaction (PCR) products was used to verify the mutation and to screen additional patients and controls.
Reverse-transcriptase PCR
Retinal RNA was isolated from human retinas using TRI Reagent (Sigma-Aldrich, St Louis, MO), and RNA derived from different human tissues was purchased (Clontech, Mountain View, CA, USA; cat. 636643, lot 8101369A). Complementary DNA was synthesized using the Verso cDNA kit (Thermo Fisher Scientific, Waltham, MA, USA) in accordance with the manufacturer’s protocol. The PCR-specific primers for arylsulfatase G (ARSG) and an internal control (PGM1) are listed in Supplementary Table S3 online.
Lysosomal enzymatic activities
The lysosomal activities in plasma and leukocytes were determined using standard fluorimetric methods (as described by Z. Lukacs in Laboratory Guide to the Methods in Biochemical Genetics (N. Blau, M. Duran, and K.M. Gibson, eds), Springer: Berlin, Germany, 2008:287–324).
Mutagenesis and cloning
The ARSG-D45Y mutant was generated by site-directed mutagenesis using a pCI-neo hARSG-WT construct as template encoding human wild-type ARSG equipped with a C-terminal RGS-His6 tag.15 PCR was conducted using Kapa HiFi polymerase (Kapa Biosystems, Wilmington, MA, USA) and the primers 5′-gtgattattttggccgattacatggggtggggtg-3′ and 5′-caccccaccccatgtaatcggccaaaataatcac-3′, respectively. All plasmids were controlled by Sanger sequencing of the ARSG insert.
Generation and cultivation of stable cell lines
HT1080 fibrosarcoma cells were transfected with the pCI-neo hARSG-D45Y plasmid using PEI (Polysciences Europe GmbH, Hirschberg an der Bergstrasse, Germany) in OptiMEM (Gibco, Thermo Fisher Scientific, Waltham, MA, USA). Stably transfected cell clones were selected by adding 800 μg/ml Geniticin (G-418) for 14 days of culture. Clones were screened for ARSG expression by immunoblotting using anti-RGS-His6 (Qiagen, Hilden, Germany), anti-ARSG (R&D Systems, Minneapolis, MN, USA) and anti-GAPDH Santa Cruz, Santa Cruz, Dallas, TX, USA; Invitrogen, Carlsbad, CA, USA) antibodies together with corresponding HRP-coupled secondary antibodies (Invitrogen, Carlsbad, CA, and Dianova, Hamburg, Germany). The cell lines expressing ARSG wild type and the ARSG-C84A mutant had been generated in a previous study.15 Cells were grown in Dulbecco’s Modified Eagle Medium (Invitrogen) containing 10% FCS (PAN), 2 mM glutamine (Lonza, Basel, Switzerland), and penicillin/streptomycin (Lonza, Basel, Switzerland) under standard culture conditions (37 °C, 5% CO2).
Sulfatase activity quantification
Cell lysates were prepared in ice-cold lysis buffer (TBS with 1 mM PMSF, 5 mM IAA, 1 mM EDTA and 0.1% (v/v) Triton X-100). For this purpose, cell pellets were resolved in lysis buffer and sonified (3 × 20 s, 40% intensity, 4 °C, Branson Sonifier, Slough, UK). The lysates were cleared by centrifugation at 18,000g at 4 °C for 15 min, and protein concentration was determined using the DC Protein Assay (Bio-Rad, Hercules, CA) with bovine serum albumen as a standard. Sulfatase activity was measured using the pseudosubstrate paranitrocatechol sulfate (Sigma-Aldrich Chemie Gmbh, Munich, Germany) as described previously.16 Specific activities were calculated and normalized according to western blot signals, i.e., after ARSG quantification by SDS-PAGE and immunoblotting using polyvinylidene fluoride membranes and the antibodies listed above. Band intensities were determined using ImageJ. Statistical analyses were conducted with GraphPad Prism (GraphPad Software, La Jolla, CA).
Molecular modeling and structural analysis
The ARSG structure was modeled using SwissModel with the structures of human arylsulfatase A (ARSA) (PDB 1e1z) or a catalytic formylglycine (FGly)-ester of human ARSA (PDB 1n2k) as templates. The ARSG and ARSA structures were visualized and manipulated using PyMol (DeLano Scientific, Palo Alto, CA).
Results
Clinical description of recruited patients
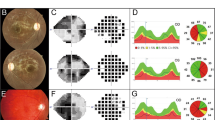
In our cohort of more than 1,500 Israeli and Palestinian families with IRDs, five affected individuals from three families shared the same ethnicity (Yemenite Jewish) and presented with a particular combination of late-onset IRD and SNHL. While some variability was present in the clinical manifestations among subjects (detailed case reports are provided in Supplementary Information Appendix), a characteristic pattern could be identified: all affected individuals presented with a distinctive retinal phenotype, including ring-shaped retinal atrophy delimiting the vascular arcades temporally and extending beyond the optic nerve nasally, with relative preservation of the mid- and far-periphery. Over time, pigment migration occurred within the atrophic areas, forming bone spicule–like pigmentary changes as well as pigment clumps, and the central macula also became involved (Figure 1). Fundus autofluorescence imaging using the Optos ultrawide-field retinal camera emphasized the areas of involvement, showing the hypofluorescent ring of atrophy surrounded by hyperfluorescent borders (Figure 1). On vertical and horizontal optical coherence tomography imaging, retinal thinning with loss of the outer retinal layers was evident also within the macular area, including the fovea at later ages (Figure 1). Functionally, Goldmann visual fields correlated with the structural changes, showing a central ring scotoma with relative preservation of central vision at earlier stages of disease. Later on, this progressed to involve the macular area with development of central scotomas, while mid- and far-peripheral visual fields remained relatively preserved (Supplementary Figure S1). Electroretinographic testing showed severely decreased rod and mixed cone–rod responses (Table 1). Electrooculography testing was performed in three affected individuals and the Arden ratio was found to be reduced (Table 1). These findings suggest injury to the retinal-pigmented epithelium, either as a primary event or secondary to photoreceptor degeneration. With regard to involvement of the auditory system, all affected subjects presented with progressive moderate to severe SNHL at a relatively late age (usually after the age of 40 years—Supplementary Figure S2 online and Supplementary Table S1). None of the affected individuals reported significant abnormalities of the vestibular system (detailed description in Supplementary Information). In addition, the following clinical evaluations and tests performed in the affected individuals were all within normal limits: lack of neurological abnormalities (all affected individuals), normal brain magnetic resonance imaging (performed on one patient), normal abdominal ultrasound exam (performed in two of the patients), and normal biochemical blood and urine tests (four patients). Osteoporosis, as determined by bone density measurements, is present in one of the patients and osteopenia in the two sisters of family MOL0737. It should be noted that at the time of all the above testing the patients were of middle age or beyond, between the ages of 50 and 72.

(a–g) MOL0737 II:1 (56 years), (h–n) MOL0737 II:2 (55 years), (o and p) MOL0120 III:2 (72 years), (q–v) MOL0120 III:1 (69 years). a, h, o, s and b, i, p, t represent ultrawide-field pseudocolor and FAF photos, respectively, taken using the Optos Panoramic 200 Optomap Fundus Camera. Characteristic ring-shaped atrophic changes accompanied by intraretinal pigment migration along the major vascular arcades and extending beyond the optic disc can be seen. (e and l) Goldmann visual field of the parallel fundus photos (a and h), respectively, each showing the corresponding ring-shaped scotoma. (c, d, j, and k) Heidelberg Spectralis multicolor fundus photos and FAF of the posterior pole of subjects MOL0737 II:1 and MOL0737 II:2, respectively. (q and r) Spectralis FAF images of the posterior pole of subject MOL0120 III:1, demonstrating macular involvement. (f, g, m, n, u, and v) are optical coherence tomography sections showing retinal thinning and loss of the outer retinal layers in the macular area of subjects MOL0737 II:1, MOL0737 II:2 and MOL0120 III:1, respectively.
Genetic analysis of study participants
Aiming to identify the cause of disease in these families, we initially performed homozygosity mapping on four individuals (MOL0737 II:1, MOL0737 II:2, MOL0120 III:1, and TB55 II:1—Figure 2a), using single-nucleotide polymorphism arrays. Following analysis with HomozygosityMapper, family MOL0737 revealed two large homozygous regions (a 9.3 Mb region on chr. 5: 126.2–135.5 Mb and a 4-Mb region on chr. 17: 66–70 Mb), shared by the two affected sisters, neither of which included a known USH gene or a clear-cut candidate for the disease. Homozygosity mapping of MOL0120 III:1 revealed four large homozygous regions (more than 20 Mb), one of which on chr. 17 (50.4–76.4 Mb) showed overlap with the one identified in family MOL0737. Patient TB55 II:1 harbored eight homozygous regions of over 10 Mb each, one of which was a 12.58-Mb homozygous region on chr. 17 (56.8–69.4 Mb). All four patients had a 3.4-Mb identical haplotype on chr. 17 (66–69.4 Mb). We subsequently performed WES analysis on three samples (MOL0737 II:2, MOL0120 III:1, and TB55 II:1, Figure 2a) and WGS on one sample (MOL0737 II:1), and all were negative for suspected disease-causing mutations in any of the 226 known IRD genes.17 WES and WGS data were also analyzed for homozygosity and the combined information revealed a single shared homozygous region on chr. 17 (66–69.4 Mb). Variant analysis revealed a single shared missense transversion, c.133G>T, p.D45Y (chr.17: 66303768G>T), in exon 2 of ARSG (NM_014960.4), which was validated by Sanger sequencing (Supplementary Figure S3) and found to be in a homozygous state in all five affected individuals. All haplotypes surrounding the variant site were identical and this variant is therefore a founder mutation.

(a) Pedigrees of families with the ARSG mutation. Filled symbols designate affected individuals. Red arrows represent index cases. Family number is depicted above each family tree and individual number below each symbol. The ARSG genotype of recruited individuals is depicted below the individual number: M, c.133G>T. Applied sequencing techniques are presented as acronyms above each subject. (b) Multiple alignment of the amino acid sequence flanking the ARSG-p.D45 mutated amino acid shows complete preservation of the aspartate in different species. (c) Multiple alignment of the amino acid sequence flanking the mutated amino acid shows preservation of the relevant aspartate in 16 of the 17 human members of the sulfatase family. (d) A WebLogo image showing the relative preservation of amino acids around ARSG-p.D45 in 74 related sequences. (e) Modeling of the human ARSG active site. The structure of human ARSG was modeled using human arylsulfatase A (ARSA), the closest homologue of ARSG, as a template (PDB code 1e1z). The model is presented as surface structure. The green color indicates the catalytic Cα-formylglycine (FGly) residue, which is located next to a cavity in the ARSG surface. Mutation of aspartate-45 (magenta) to tyrosine (red) positions the phenolic ring of the tyrosine within this cavity, thereby altering the accessibility of the FGly and interfering with substrate desulfation. (f) Molecular structure of the calcium-binding site of ARSA. (Left) The four complexing aspartate and asparagine residues are shown in white with heteroatoms shown in red and blue. The FGly residue is shown in green. The complexed metal ion is represented by the green sphere. In this structure (PDB code 1n2k) a critical intermediate of the catalytic cycle was trapped using 4-methylumbeliferyl phosphate as a substrate surrogate which leads to FGly phosphorylation, thereby mimicking the physiological FGly sulfation. The phosphor atom is shown in orange. (Right) Upon D30Y mutation the tyrosine side chain clashes with the phosphate group, indicating sterical hindrance of substrate binding and, consequently, of covalent FGly-ester formation. In addition, the coordination of the metal ion will be massively distorted.
To rule out the possibility that the ARSG c.133G>T variant is a polymorphism in the Yemenite Jewish population, we screened 101 controls and identified one heterozygous individual (this corresponding to a minor allele frequency of 0.005). In addition, the variant does not appear in any of the available databases, including gnomAD (with 246,228 sequenced alleles of various origins). We subsequently used Sanger sequencing of exon 2 to screen the remaining unsolved IRD index cases of Yemenite origin (n = 19) for p.D45Y, as well as other mutations in this exon. The analysis revealed a single heterozygous case with early-onset RP and normal hearing at the age of 46 years. Sequencing analysis of all ARSG exons failed to detect a second possible pathogenic variant in this individual, and we therefore predict that she carries p.D45Y by chance, while the disease-causing mutation lies in a yet-to-be-identified RP-causing gene. The genotype difference between the cohort of IRD families (three homozygous: one heterozygous: 18 wt) and controls (none homozygous: one heterozygous: 100 wt) of Yemenite Jewish origin is statistically significant (p < 4.0 × 10−4). Aiming to expand the ARSG tissue expression profile,18 we performed reverse-transcriptase PCR analysis of 17 different human tissues, which revealed a ubiquitous pattern, with ARSG being expressed in the majority of the studied tissues, including the retina (Supplementary Figure S4).
p.D45 is critical for ARSG function
In the encoded arylsulfatase G (ARSG) enzyme, an ensemble of ten conserved amino acid residues forms the catalytic pocket, as holds true for type I sulfatases in general.18,19 For ARSG, one of these ten residues is aspartate at position 45. This amino acid is therefore extremely important and did not tolerate any substitution in the course of evolution (Figure 2b–d). It is perfectly conserved not only among all known ARSG orthologues (Figure 2d, which is based on 74 ARSG-related sequences) but also among 16 of the 17 members of the human sulfatase family (Figure 2c). Of note, mutations in the corresponding aspartate residue in N-sulfoglucosamine sulfohydrolase (encoded by SGSH) and N-acetylgalactosamine-6-sulfatase (encoded by GALNS) cause loss-of-function alleles resulting in different forms of mucopolysaccharidosis (MPS type IIIA and IVA, respectively).20,21
Arylsulfatase G was identified in 2002 as a novel sulfatase gene18 and has been further characterized biochemically as a lysosomal sulfatase.15 As mentioned above, aspartate-45 is part of the ARSG active site, where it coordinates a calcium ion next to the catalytic formylglycine (FGly) residue that is essential for all human sulfatases.22Figure 2e shows the surface structure of the ARSG active site, as modeled on the known structure of human ARSA, i.e., the closest relative of ARSG (see Figure 2c). A tyrosine (Figure 2e, red) replacing the aspartate (magenta), as found in the patients described here, fills part of the active site cavity next to the FGly (green). The bulky tyrosine side-chain leaves no space for formation of the FGly-sulfate ester, which is an essential intermediate of the catalytic cycle,23 as shown in Figure 2f on the basis of a published crystal structure with a trapped ester intermediate in ARSA.24 In addition, a loss of the calcium ion may be predicted as a consequence of a massively distorted coordination, because the tyrosine hydroxyl at a distance of 4.0 Å is too remote (2.4 Å for the wild-type aspartate carboxyl group). Loss of the calcium ion in turn may also affect protein stability.
ARSG-p.D45Y abolishes enzyme activity but does not lead to significant glycosaminoglycan excretion in patients
To test these predictions experimentally, ARSG was stably expressed in human fibrosarcoma HT1080 cells, one of the most efficient cell lines used for production of human sulfatases.15,25 Three independent clones expressing the p.D45Y-mutant were compared to wild-type ARSG, as well as to the null-mutant ARSG-p.C84A (Figure 3), which lacks the catalytic FGly-84 (deriving from the posttranslational modification of Cys84).26 No significant sulfatase activity was detectable in the p.D45Y clones (Figure 3a, b), despite the fact that all ARSG variants were expressed at similar levels (Figure 3c). The measured residual activity (<20% of ARSG wild type) was in fact as low as that observed for the p.C84A null mutant (Figure 3a) and can be attributed to the endogenous sulfatase activity present in the HT1080 cells.15 In addition to this marked impairment of enzymatic activity of ARSG-p.D45Y, we observed lack of processing of the ARSG precursor protein (63 kDa) into its mature three chains, of which the largest subunit (~30 kDa in humans),16 could be detected only for the wild-type protein (Figure 3d). Lack of processing was observed earlier also for ARSG-p.C84A (unpublished data) and also for active site mutants of two other human sulfatases.27,28 While this maturation of ARSG is dispensable for enzymatic activity in vitro, the observed lack of in vivo processing, which relies on the lysosomal proteases cathepsin B and L,16 suggests that ARSG-p.D45Y is improperly transported to the lysosomes. To conclude, the p.D45Y-mutation, apart from blocking the catalytic activity, clearly affects maturation and probably also trafficking of the enzyme to its target organelle. However, a drastic decrease in stability of the ARSG-p.D45Y precursor protein was not observed (Figure 3c).

(a and b) Cell lysates of HT1080 cells, stably overexpressing ARSG wild type, the null-mutant C84A, or the D45Y mutant (independent cell clones D45Y a, D45Y b and D45Y c), were subjected to sulfatase activity assays using the chromogenic pseudosubstrate para-nitrocatechol sulfate. The enzyme reaction was terminated after 60 min (a) or after 30, 60, and 240 min (b), as indicated. The given specific activities were normalized according to western blot signals, using an anti-RGS-His6 antibody (c) to correct for differences in mutant expression. These corrections explain the apparently lower activity for the high-expressing D45Y clones a and b, as compared to D45Y clone c and the C84A null-mutant, all of which in fact show only background activities owing to endogenous sulfatases. Error bars represent standard deviation from three experiments. (d) Analysis of ARSG processing. Cell lysates of overexpressing cells were blotted and ARSG was detected using an anti-ARSG antibody. The processed ~30 kDa form of mature ARSG was observed only for ARSG-WT. GAPDH detection served as loading control (c and d).
Since the ARSG-KO in the mouse model and also mutations in genes that are paralogous to ARSG cause phenotypes affecting additional lysosomal enzymes and catabolic pathways,29,30 we tested patient samples for biochemical parameters that might be affected by ARSG deficiency (Supplementary Table S2). However, the enzymatic activities of β-galactosidase, total hexosaminidase, and arylsulfatase A in plasma samples of two patients, as well as α-L-iduronidase and β-galactosidase in leukocytes, were all within normal ranges. Of note, urinary glycosaminoglycans were at the upper limit of the normal range (Supplementary Table S2).
Discussion
We report here that a mutation in a gene encoding a lysosomal protein, ARSG, causes atypical USH in humans. Patients homozygous for the p.D45Y variant manifest a late-onset rod-cone dystrophy with development of atrophy and pigmentary changes encroaching the posterior pole and causing a ring scotoma, which later progresses to involve the macula and fovea while the mid- and far-periphery of the retina are relatively preserved. This is accompanied by progressive moderate to severe SNHL.
In other species, mutated canine ARSG on the basis of its symptomatic presentation has been reported to cause late-onset neuronal ceroid lipofuscinosis (NCL),31 a neurodegenerative lysosomal storage syndrome that includes static and dynamic ataxia and cerebellar atrophy. Although NCL in other models usually also triggers retinal degeneration (but not hearing loss), none of the 138 studied dogs showed visual impairment. In mice, Arsg knockout showed that the protein in fact is responsible for the degradation of 3-O-sulfated N-sulfoglucosamine residues of heparan sulfate glycosaminoglycans, causing a new form of lysosomal storage disease, which was termed MPS IIIE.32,33 These mice were shown to also have photoreceptor degeneration.34 In this context, it should be mentioned that retinal degeneration is a very common finding in many human lysosomal storage diseases, including NCL and MPS. Remarkably, photoreceptor degeneration in Arsg-knockout mice appeared clearly before manifestation of symptoms affecting the central nervous system, including Purkinje cell loss and ataxia. The USH patients described here did not show any neurological symptoms or signs of peripheral lysosomal storage, even at a relatively old age (the oldest patient described here is 72 years old), but we cannot rule out the possibility that mild and very late-onset symptoms may appear. The phenotype in humans indicates that, as in mice, the retina is among the primary tissues affected by ARSG deficiency, suggesting that 3-O-sulfated heparan sulfate has an important physiological function in the retina and the inner ear. Nevertheless, it should also be mentioned that in both NCL (type CLN7) and MPS (type IIIC), diseases that are generally fatal in late adolescence or early adulthood,35,36 an increasing number of patients were recently described as expressing isolated RP, while the severe neurological and peripheral somatic features that are typically observed in both diseases were lacking.37,38,39 In this respect, it should also be noted that the (almost) normal level of urinary glycosaminoglycans in our patients is not in accordance with findings among classical MPSs. Thus, ARSG patients are likely to be missed in the first-line screening for MPS diseases. Interestingly, also other MPS III cases were occasionally reported who presented with urinary glycosaminoglycan levels in the normal range.40
The phenotype which we describe here differs from the three accepted USH types that were defined historically, and adds to the large number of “atypical” USH cases that has emerged in recent years. This may further emphasize the need to update the nomenclature of USH syndrome, in order to better distinguish between the different USH subtypes.
References
Boughman JA, Vernon M, Shaver KA. Usher syndrome: definition and estimate of prevalence from two high-risk populations. J Chronic Dis 1983;36:595–603.
Rosenberg T, Haim M, Hauch AM, Parving A. The prevalence of Usher syndrome and other retinal dystrophy-hearing impairment associations. Clin Genet 1997;51:314–321.
Kimberling WJ, Hildebrand MS, Shearer AE et al. Frequency of Usher syndrome in two pediatric populations: implications for genetic screening of deaf and hard of hearing children. Genet Med. 2010;12:512–516.
Khateb S, Zelinger L, Mizrahi-Meissonnier L et al. A homozygous nonsense CEP250 mutation combined with a heterozygous nonsense C2orf71 mutation is associated with atypical Usher syndrome. J Med Genet 2014;51:460–469.
Bashir R, Fatima A, Naz S. A frameshift mutation in SANS results in atypical Usher syndrome. Clin Genet 2010;78:601–603.
Liu XZ, Hope C, Walsh J et al. Mutations in the myosin VIIA gene cause a wide phenotypic spectrum, including atypical Usher syndrome. Am J Hum Genet 1998;63:909–912.
Pierrache LHM, Kimchi A, Ratnapriya R et al. Whole-exome sequencing identifies biallelic IDH3A variants as a cause of retinitis pigmentosa accompanied by pseudocoloboma. Ophthalmology 2017;124:992–1003.
Gong B, Wei B, Huang L et al. Exome sequencing identified a recessive RDH12 mutation in a family with severe early-onset retinitis pigmentosa. J Ophthalmol 20152015: 942740.
Nishiguchi KM, Tearle RG, Liu YP et al. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc Natl Acad Sci USA 2013;110:16139–16144.
Carss KJ, Arno G, Erwood M et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am J Hum Genet 2017;100:75–90.
Kremer H, van Wijk E, Marker T, Wolfrum U, Roepman R. Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum Mol Genet 2006;15 Spec No 2:R262–270.
Reiners J, Nagel-Wolfrum K, Jurgens K, Marker T, Wolfrum U. Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp Eye Res 2006;83:97–119.
El-Amraoui A, Petit C. The retinal phenotype of Usher syndrome: pathophysiological insights from animal models. C R Biol 2014;337:167–177.
Flanagan AM, Stavenschi E, Basavaraju S, Gaboriau D, Hoey DA, Morrison CG. Centriole splitting caused by loss of the centrosomal linker protein C-NAP1 reduces centriolar satellite density and impedes centrosome amplification. Mol Biol Cell 2017;28:736–745.
Frese MA, Schulz S, Dierks T. Arylsulfatase G, a novel lysosomal sulfatase. J Biol Chem 2008;283:11388–11395.
Kowalewski B, Lubke T, Kollmann K et al. Molecular characterization of arylsulfatase G: expression, processing, glycosylation, transport, and activity. J Biol Chem 2014;289:27992–28005.
Beryozkin A, Shevah E, Kimchi A et al. Whole exome sequencing reveals mutations in known retinal disease genes in 33 out of 68 Israeli families with inherited retinopathies. Sci Rep 2015;5:13187.
Ferrante P, Messali S, Meroni G, Ballabio A. Molecular and biochemical characterisation of a novel sulphatase gene: arylsulfatase G (ARSG). Eur J Hum Genet 2002;10:813–818.
Waldow A, Schmidt B, Dierks T, von Bulow R, von Figura K. Amino acid residues forming the active site of arylsulfatase A. Role in catalytic activity and substrate binding. J Biol Chem 1999;274:12284–12288.
Beesley CE, Young EP, Vellodi A, Winchester BG. Mutational analysis of Sanfilippo syndrome type A (MPS IIIA): identification of 13 novel mutations. J Med Genet 2000;37:704–707.
Morrone A, Tylee KL, Al-Sayed M et al. Molecular testing of 163 patients with Morquio A (mucopolysaccharidosis IVA) identifies 39 novel GALNS mutations. Mol Genet Metab 2014;112:160–170.
Schmidt B, Selmer T, Ingendoh A, von Figura K. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell 1995;82:271–278.
Boltes I, Czapinska H, Kahnert A et al. 1.3 A structure of arylsulfatase from Pseudomonas aeruginosa establishes the catalytic mechanism of sulfate ester cleavage in the sulfatase family. Structure 2001;9:483–491.
Chruszcz M, Laidler P, Monkiewicz M, Ortlund E, Lebioda L, Lewinski K. Crystal structure of a covalent intermediate of endogenous human arylsulfatase A. J Inorg Biochem 2003;96:386–392.
Dierks T, Schmidt B, Borissenko LV et al. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell 2003;113:435–444.
Dierks T, Lecca MR, Schlotterhose P, Schmidt B, von Figura K. Sequence determinants directing conversion of cysteine to formylglycine in eukaryotic sulfatases. EMBO J 1999;18:2084–2091.
Daniele A, Di Natale P. Heparan N-sulfatase: cysteine 70 plays a role in the enzyme catalysis and processing. FEBS Lett 2001;505:445–448.
Millat G, Froissart R, Maire I, Bozon D. Characterization of iduronate sulphatase mutants affecting N-glycosylation sites and the cysteine-84 residue. Biochem J. 1997;326(Pt 1):243–247.
Van Hoof F, Hers HG. The abnormalities of lysosomal enzymes in mucopolysaccharidoses. Eur J Biochem 1968;7:34–44.
Vitner EB, Platt FM, Futerman AH. Common and uncommon pathogenic cascades in lysosomal storage diseases. J Biol Chem 2010;285:20423–20427.
Abitbol M, Thibaud JL, Olby NJ et al. A canine arylsulfatase G (ARSG) mutation leading to a sulfatase deficiency is associated with neuronal ceroid lipofuscinosis. Proc Natl Acad Sci USA 2010;107:14775–14780.
Kowalewski B, Heimann P, Ortkras T et al. Ataxia is the major neuropathological finding in arylsulfatase G-deficient mice: similarities and dissimilarities to Sanfilippo disease (mucopolysaccharidosis type III). Hum Mol Genet 2015;24:1856–1868.
Kowalewski B, Lamanna WC, Lawrence R et al. Arylsulfatase G inactivation causes loss of heparan sulfate 3-O-sulfatase activity and mucopolysaccharidosis in mice. Proc Natl Acad Sci USA 2012;109:10310–10315.
Kruszewski K, Lullmann-Rauch R, Dierks T, Bartsch U, Damme M. Degeneration of photoreceptor cells in arylsulfatase G-deficient mice. Invest Ophthalmol Vis Sci 2016;57:1120–1131.
Kousi M, Siintola E, Dvorakova L et al. Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain 2009;132(Pt 3):810–819.
Wraith JEMucopolysaccharidoses and oligosaccharidoses. In: Fernandes J, Saudubray JM, van den Berghe G, Walter JH(eds). Inborn Metabolic Diseases. Diagnosis and Treatment. Vol 4. Springer: Heidelberg, Germany, 2006: 496–507.
Khan KN, El-Asrag ME, Ku CA et al. Specific alleles of CLN7/MFSD8, a protein that localizes to photoreceptor synaptic terminals, cause a spectrum of nonsyndromic retinal dystrophy. Invest Ophthalmol Vis Sci 2017;58:2906–2914.
Roosing S, van den Born LI, Sangermano R et al. Mutations in MFSD8, encoding a lysosomal membrane protein, are associated with nonsyndromic autosomal recessive macular dystrophy. Ophthalmology 2015;122:170–179.
Haer-Wigman L, Newman H, Leibu R et al. Non-syndromic retinitis pigmentosa due to mutations in the mucopolysaccharidosis type IIIC gene, heparan-alpha-glucosaminide N-acetyltransferase (HGSNAT). Hum Mol Genet 2015;24:3742–3751.
Giugliani RThe mucopolysaccharidoses. In: Metha A, Winchester B(eds). Lysosomal Storage Disorders: A Practical Guide. John Wiley: Chichester, UK, 2012: 94–100.
Acknowledgments
This study was financially supported by the Foundation Fighting Blindness USA (BR-GE-0214-0639 to D.S., E.B., and T.B.-Y.), Israel Science Foundation (2154/15 to S.K.), Chief Scientist Office of the Israeli Ministry of Health and the Lirot association (300009177 to S.K. and 300011893 to D.S. and T.B.-Y.), the Yedidut Research Grant (to E.B.), the Deutsche Forschungsgemeinschaft (DI 575/10-1 to T.D.), and the Swiss National Science Foundation (156260 to C.R.). The authors thank all patients and family members for their participation in this study, and Torben Lübke for critically reading the manuscript. Geto Mengisto, Kerstin Fentker and Mai-Britt Ilse, Inbar Erdinest, and Devora Marks-Ohana are acknowledged for expert technical assistance.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
The authors declare no conflict of interest
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Khateb, S., Kowalewski, B., Bedoni, N. et al. A homozygous founder missense variant in arylsulfatase G abolishes its enzymatic activity causing atypical Usher syndrome in humans. Genet Med 20, 1004–1012 (2018). https://doi.org/10.1038/gim.2017.227
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.227
Keywords
This article is cited by
-
Exploring the support needs of Australian parents of young children with Usher syndrome: a qualitative thematic analysis
Orphanet Journal of Rare Diseases (2024)
-
The genetic and phenotypic landscapes of Usher syndrome: from disease mechanisms to a new classification
Human Genetics (2022)
-
Usher syndrome type IV: clinically and molecularly confirmed by novel ARSG variants
Human Genetics (2022)
