
Abstract
Purpose
Combined pituitary hormone deficiency (CPHD) is characterized by a malformed or underdeveloped pituitary gland resulting in an impaired pituitary hormone secretion. Several transcription factors have been described in its etiology, but defects in known genes account for only a small proportion of cases.
Methods
To identify novel genetic causes for congenital hypopituitarism, we performed exome-sequencing studies on 10 patients with CPHD and their unaffected parents. Two candidate genes were sequenced in further 200 patients. Genotype data of known hypopituitary genes are reviewed.
Results
We discovered 51 likely damaging variants in 38 genes; 12 of the 51 variants represent de novo events (24%); 11 of the 38 genes (29%) were present in the E12.5/E14.5 pituitary transcriptome. Targeted sequencing of two candidate genes, SLC20A1 and SLC15A4, of the solute carrier membrane transport protein family in 200 additional patients demonstrated two further variants predicted as damaging. We also found combinations of de novo (SLC20A1/SLC15A4) and transmitted variants (GLI2/LHX3) in the same individuals, leading to the full-blown CPHD phenotype.
Conclusion
These data expand the pituitary target genes repertoire for diagnostics and further functional studies. Exome sequencing has identified a combination of rare variants in different genes that might explain incomplete penetrance in CPHD.
Similar content being viewed by others


Introduction
The pituitary gland is the master endocrine gland of the body, regulating growth, metabolism, stress response, reproduction, and homeostasis. The mature organ is composed of two structures derived from different ectodermal origins: the adenohypophysis (anterior pituitary, consisting of the anterior and intermediate lobes) and the neurohypophysis (posterior lobe). The anterior pituitary secretes several hormones—growth hormone, thyroid-stimulating hormone, follicle-stimulating hormone, luteinizing hormone, adrenocorticotrophic hormone, and prolactin—from five different cell types that all originate from a single primordial structure, Rathke’s pouch. The development of the anterior pituitary has been extensively studied in mice and is highly conserved across vertebrates, including humans.1,2
In mice it begins with a thickening of the oral ectoderm at E8.5, forming a definitive Rathke’s pouch at E12.5 as it comes into contact with the infundibulum, an evagination from the ventral diencephalon that will form the future posterior lobe. From E12.5 to E17.5, progenitor cells proliferate and finally differentiate into the specified hormone-secreting cell types. The adult anterior gland, which is completely separated from the oral cavity, is composed of the dorsal intermediate lobe and the ventral anterior lobe.
Several pathways, including Bmp, Shh, Fgf, Wnt, and Notch signaling, orchestrate the sequential expression of transcription factors and pituitary-relevant genes crucial for Rathke’s pouch development, including initiation, regional cell identity, and proliferation, as well as final cellular differentiation into the five hormone-secreting cell types. Abnormalities within these processes can result in the reduction or loss of hormone-secreting cells causing congenital hypopituitarism. Clinical features of hypopituitarism are variable; the key symptoms are growth retardation, developmental delay, delayed puberty and infertility, poor stress response, and metabolic abnormalities such as hypoglycemia. In addition to pituitary abnormalities (aplasia or hypoplasia of the anterior pituitary, ectopic posterior pituitary, absent infundibulum), patients can show eye abnormalities or midline defects.3,4,5 Mutations in some genes—those encoding the transcription factors PROP1, GLI2, LHX3, LHX4, HESX1, PITX2, SOX2, SOX3, OTX2, POU1F1, and TBX19—have already been reported in the etiology of congenital hypopituitarism in humans, resulting in either loss of one pituitary hormone (isolated pituitary hormone deficiency), deficiency of two or more hormones (combined pituitary hormone deficiency, CPHD), or syndromic disorders such as septo-optic dysplasia.1,5,6 CPHD has an estimated prevalence of 1 in 8,000 individuals worldwide (https://ghr.nlm.nih.gov/condition/combined-pituitary-hormone-deficiency). Most cases of CPHD are sporadic; familial inheritance accounts for 5 to 30% of all cases.7 From all genes implicated in the etiology of CPHD, PROP1 is reported as the most common genetic cause.1,5,8,9,10,11,12 Yet defects within the known genes only account for a small percentage of CPHD cases, suggesting that many of the underlying genes involved in the etiology of CPHD still have to be identified.13 In this study we aimed to find novel genetic causes of congenital hypopituitarism in a cohort of 210 patients by performing trio-based exome sequencing on 10 of the patients as a starting point. Following several filtering steps, a list of candidate genes was compiled. Two candidate genes were analyzed in a cohort of 200 patients in more detail, and two further variants in SLC20A1 and SLC15A4 were identified. In addition, transmitted rare, likely damaging variants from known CPHD genes were detected.
Materials and methods
Patients and controls
For exome sequencing, 10 CPHD patients (2 females and 8 males) from the Center of Child and Youth Medicine in Heidelberg and their healthy parents were recruited with their informed consent. All patients were diagnosed between the ages of 1 week and 4 years and showed growth hormone and thyroid-stimulating hormone deficiency. Additionally, adrenocorticotrophic hormone and/or follicle-stimulating hormone/luteinizing hormone deficiencies were detected in 9 and 7 patients, respectively. None of the patients showed a prolactin deficiency, measured via immunoassay (basal and/or after thyrotropin-releasing hormone stimulation). Magnetic resonance imaging revealed abnormal brain morphologies, including aplasia or hypoplasia of the adenohypophysis (all cases), ectopic neurohypophysis (9 cases), and an aplastic infundibulum (7 cases). The sella turcica was aplastic in one patient and the nervus opticus hypoplastic in another. DNA was extracted from peripheral blood leukocytes using the salting-out method. In total, 30 (patients and parents) samples were exome-sequenced.
For targeted sequencing, DNA from 200 patients was provided by R.W.P., with their informed consent. Twenty-four percent of these patients presented the refined subphenotype CPHD plus ectopic posterior pituitary. Sequencing data from the Exome Variant Server (the National Heart, Lung, and Blood Institute’s “Grand Opportunity” Exome Sequencing Project (ESP), Seattle, WA (http://evs.gs.washington.edu/EVS/)) and 1000 Genomes (http://www.1000genomes.org/)14 were used as controls.
Exome sequencing and filtering
The Agilent SureSelect Human All Exon V4 (without untranslated regions) was used to capture the exons. Sequencing was done using the Illumina HiSeq 2000 system (Illumina GmbH, Münschen, Germany). The raw sequence data were mapped to the 1000 Genomes phase II reference genome (hs37d5) using Burrows–Wheeler Aligner 0.6.2, and the duplicates were removed using PICARD. The 30 samples showed an average exon coverage of 95.872x and a median-of-median exon coverage of 90.32x. 99.904% of exons showed non-zero coverage and 99.252% of exons had at least 10x coverage. On average, the bases in the target regions with QUAL > 1 showed a coverage of 105.575x with 99.026% showing a minimum of 10x coverage and 97.068% a minimum 20x coverage. The single-nucleotide variants (SNVs) were called by SAMtools on individual samples and the respective parent information was merged later. Indels were then called along with parent samples using Platypus. In-house pipelines were used to annotate the variants with two databases: the National Center for Biotechnology Information’s dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP) and 1000 Genomes. ANNOVAR and RefSeq (downloaded on 20 February 2011) were used for the functional annotation of the variants.
For further analysis, SNVs and indels with a read depth of at least 10 × and minimum QUAL score of 20 were considered. Nonsynonymous (missense, stop gain, stop loss) and splice site–affecting SNVs as well as all exonic indels were filtered further. Custom variant allele frequency (VAF) thresholds were used to assign genotypes to the SNVs. For SNVs to be heterozygous, they should have a minimum of 15% VAF and a maximum of 90% VAF. SNVs with VAF of 95% and above were considered homozygous alternate and those with VAF of 5% and below were considered homozygous reference. For indels, the genotype predicted by Platypus was used.
The minor allele frequency (MAF) from 1000 Genomes was used to define the variant’s rareness in the population. Variants with MAF >1% were removed (removal of compound heterozygous variants if MAF of one allele >1%). Furthermore, a set of 45 exomes from the in-house database was used as control to remove common variants and sequencing artifacts (removal of compound heterozygous variants if one or both alleles was found in in-house controls).
The functional effect of the remaining variants was predicted using the in silico prediction tools MutationTaster, SIFT, PolyPhen2, PROVEAN, and CADD. Variants that were predicted as damaging by at least one in silico prediction tool were used for further analysis. Compound heterozygous variants were included only if both variants are predicted as damaging by at least one prediction tool.
Pituitary complementary DNA libraries
Data for two complementary DNA (cDNA) libraries generated from embryonic mouse Rathke’s pouch material are publicly available from the National Center for Biotechnology Information (UniGene Library Browser).6,15
E12.5: UniGene, RIKEN full-length enriched mouse cDNA library, Rathke’s pouches 12.5 days embryo, library ID: Lib.18109
E14.5: UniGene, RIKEN full-length enriched mouse cDNA library, CD-1 Rathke’s pouches 14.5 days embryo, library ID: Lib.18113
Sanger sequencing
All variants in genes present in the cDNA libraries were confirmed by Sanger sequencing. Polymerase chain reaction was performed with Paq5000 Polymerase (Stratagene, Waldbronn, Germany) or HotStarTaq DNA Polymerase (Qiagen, Hilden, Germany). Generated polymerase chain reaction products were analyzed on agarose gels, purified, and sequenced directly using the DYEnamic ET Terminator Cycle Sequencing Kit (GE Healthcare, Rimbach, Germany) and the MegaBACE 1000 DNA Analysis System (GE Healthcare). For sequencing the coding region of SLC20A1 and SLC15A4, polymerase chain reaction products of exonic amplicons were generated using Paq5000 Polymerase (Stratagene) and subsequently sequenced (GATC, Konstanz, Germany). All primer sequences used are listed in Supplementary Table S1 online.
Pathway analysis
To identify relevant biological pathways implicating those genes identified, we applied Ingenuity Pathway Analysis (IPA) methodology (IPA Software, Ingenuity Systems, http://www.ingenuity.com). IPA integrates selected omics data sets (genomics, transcriptomics, miRNAomics, proteomics) with mining techniques to predict functional connections and their interpretation in the context of protein networks that comprise protein–protein interactions and related biological functions and canonical signaling pathways.
Results
Exome sequencing
To identify new candidate genes implicated in the etiology of congenital hypopituitarism, a cohort of 210 patients was collected. First, exome sequencing on 10 probands (2 females, 8 males; Supplementary Table S2) and their unaffected parents (total: 30 samples) was performed.
Exonic sequences were enriched using SureSelect (Agilent) for targeted exon capture and paired-end sequenced with the Illumina HiSeq 2000 system. The raw sequence data were mapped to the 1000 Genomes phase II reference genome (hs37d5) followed by a bioinformatic variant detection pipeline (see Materials and Methods and Figure 1). On average, we obtained high exon coverage of 95.872 × for all 30 samples demonstrating good quality of the sequence data. Of all exons, 99.904% were covered (nonzero coverage) and 99.252% were assessed by at least 10 independent reads.

Pipeline of exome sequencing and filtering and two approaches (a) and (b) to analyzing nontransmitted and transmitted variants. BWA, Burrows–Wheeler Aligner; SNV, single-nucleotide variant.
For our analyses only nonsynonymous (missense, stop gain, stop loss) and splice site–affecting SNVs as well as all exonic indels were filtered further. We considered SNVs and indels inherited de novo; homozygous, heterozygous, hemizygous, or compound heterozygous showing ≥10 independent reads; and a minimum QUAL score of 20. Variants annotated in 1000 Genomes with a MAF of ≤1% or found in sequence data of an in-house control group were excluded. This resulted in approximately 400 candidate genes per patient. Variants were then analyzed on their effect on protein function and were considered for further analysis if they were predicted as damaging by at least one of the prediction tools used (SNVs: PROVEAN, MutationTaster, PPH2, SIFT; indels: PROVEAN, MutationTaster, SIFT) (Figure 1a). Applying these criteria, we searched for rare highly penetrant causative variants and revealed a total number of 51 variants in 38 genes and an average number of 3.8 candidate genes (5.1 variants) for each patient (Supplementary Table S3).
IPA network analysis
To further address the biological relevance of our results, IPA was first used on all known 22 human and mouse genes underlying combined pituitary hormone from the literature. The newly identified 38 candidate genes were then integrated to predict functional connections in the context of the known protein networks. In this analysis, 6 of the 38 identified genes could be integrated in the existing networks (SLC20A1, TET2, AGAP1, BODL1, MAMLD1, and ITGA2B; from 3 patients) (Figure 2). Key nodules turned out to be the two transcription factor encoding genes, SOX2 and GATA2, and the nuclear receptor gene NR5A1.16,17 SOX2 interacts with BOD1L1, which indirectly activates AGAP1.18 SOX2 also interacts with NR5A1, which activates MAMLD119,20 and it also activates TET2, which indirectly activates AGAP1.21,22 The transcription factor GATA2 interacts with SLC20A1 and activates ITGA2B; TET2 activates not only AGAP1 but also SLC20A1.23 Together, these data link variants in several novel candidate genes with the SOX2 and GATA2 networks, which have a validated role in pituitary development and individuals with CPHD.24,25,26 Interestingly, three of the novel candidates (SLC20A1, TET2, AGAP1) are derived from patient RaKi_40; two candidates, BOD1L1 and MAMLD1, are connected with SOX2 and are derived from patient RaKi_51; and ITGA2B is from patient RaKi_42.

All known human (yellow) and mouse (green) genes from the literature underlying combined pituitary hormone deficiency and congenital pituitary malformation were analyzed using the IPA network analysis. The newly identified 38 candidate genes were added to predict functional connections in the context of the known protein networks. Six (orange) of the 38 identified candidate genes could be integrated in the existing networks.
Identification of pituitary-relevant genes
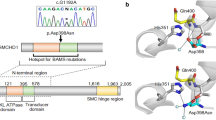
As all patients showed aplastic or hypoplastic adenohypophyses, indicating that disruption of proliferative processes during pituitary development was likely, we searched for additional evidence that the identified candidate genes were transcribed in the developing pituitary. A phase of massive cell proliferation occurs from E12.5 to E14.5 of mouse pituitary development, which prompted us to analyze the presence or absence of the 38 candidate genes in cDNA library data from mouse wild-type Rathke’s pouches at embryonic stage E12.5 and E14.5.6,15 Using this approach, a total of 11 genes, including SLC20A1 and SLC15A4, were found to be expressed at E12.5 or E14.5 or both (Table 1). All variants of these candidate genes could be confirmed by Sanger sequencing. Variants were either de novo (4 genes), or inherited homozygous (3 genes), hemizygous (3 genes), or compound heterozygous (1 gene), leading to a frameshift in one gene or to a single amino acid exchange in the remaining genes.
Sequencing SLC20A1 and SLC15A4 in a large CPHD patient cohort
Exome sequencing has discovered de novo missense variants in SLC20A1 (L89S, patient RaKi_40) and SLC15A4 (P456L, patient RaKi_54), two members of the solute carrier (SLC) group of membrane transport proteins (Supplementary Table S3, Table 2). To gain further evidence for a causative pathogenic effect in CPHD, we selected SLC20A1 and SLC15A4 as candidate genes for sequence analysis in an additional 200 patients. The two candidate genes were selected for the following reasons: (i) de novo variants, (ii) variants predicted as damaging by all used prediction tools (PPH2, PROVEAN, SIFT, MutationTaster), (iii) high CADD scores of 22.5 and 31.0, (iv) found in neither SNP (dbSNP) nor large population exome/genome sequencing databases (1000 Genomes, Exome Variant Server, and gnomAD database (http://gnomad.broadinstitute.org)), and (v) expressed in the E12.5 or E14.5 pituitary transcriptome (Table 1,Supplementary Table S3).
Sequence analysis of SLC20A1 identified one additional unrelated patient (511) with a variant resulting in an amino acid exchange from leucine to phenylalanine (L521F), which was predicted as damaging by all prediction tools (CADD score 28.9) and was absent in population databases (Table 2). Further SLC20A1 variants identified in other patients likely present nonpathogenic variants (Table 2). Sequence analysis of SLC15A4 also identified one additional unrelated patient (1801) with a variant predicted as damaging by two of the four prediction tools used (CADD score 15.9), resulting in an amino acid exchange from leucine to phenylalanine (L84F)—a variant that was absent in public population databases (e.g., the gnomAD server) (Table 2). In total, likely pathogenic variants were identified in 2 additional patients out of 200 cases analyzed, reflecting the complex nature of this disorder.
Inherited variants of known CPHD genes from asymptomatic parents
To also determine whether any variants from the known hypopituitary genes were transmitted from the unaffected parents to the affected children, we applied a second approach and went back to the initial 400 candidate genes per patient (Figure 1b). To our surprise, three patients carried likely damaging variants in the GLI2 and LHX3 genes,27,28 with CADD scores ranging between 15 and 33 (RaKi_40, RaKi_50, and RaKi_54) (Table 3). We reviewed gnomAD read-level evidence for every rare variant. Previously published LHX3 and GLI2 mutations showed CADD scores ranging between 9 and 34 for missense mutations in GLI2 and between 6 and 28 in LHX3 (Supplementary Table S4), suggesting that the variants identified in our study are also likely pathogenic. Whereas all patients with LHX3 mutations mentioned to date have been homozygous or compound heterozygous, most GLI2 variants associated to CPHD have been heterozygous with incomplete penetrance. The GLI2 variant was found in the asymptomatic father and the patient RaKi_40, but the patient also showed the de novo SLC20A1 (L89S) variant, suggesting that the added effect of the two variants led to the clinical phenotype (Supplementary Figure 1). The same was also true for the asymptomatic father of patient RaKi_54, who carried the same LHX3 variant as his affected child, but only the child additionally presented the SLC15A4 (P456L) variant. The third patient, RaKi_50, had a combination of a likely damaging GLI2 and a LHX3 variant; the GLI2 variant was transmitted from the asymptomatic father and the LHX3 variant from the asymptomatic mother (Supplementary Figure 1, Table 3).
In RaKi_51, a variant leading to a frameshift (p.S15Kfs*41) in the recently published hypopituitary gene TCF7L1 was transmitted from the unaffected mother to the affected child. Both mother and son RaKi_51 carried this rare variant, which is not annotated in the gnomAD database, strongly suggesting that it contributes to function; in the son possibly in concert with a de novo mutation in the NOBOX gene.29
Discussion
Over the past two decades our knowledge of the genes important in pituitary development mainly came from the study of animal models. In these models, defective genes encoding transcription factors and signaling molecules for organ commitment, cell differentiation, and proliferation could be identified. In humans, mutations in PROP1, POU1F1, HESX1, GLI2, LHX3 and LHX4, and others have been subsequently demonstrated in patients with CPHD, associated with severe forms of short stature, together with a remarkable variability in the overall clinical manifestation. In the majority of affected CPHD individuals, the genetic etiology has remained unexplained, suggesting the involvement of additional genes in pituitary ontogeny.
Whole-exome sequencing now provides the opportunity to identify novel genes implicated in hypopituitarism independent of preceding animal studies.30 To identify novel candidate genes associated with CPHD, we started out with high-quality exome-sequencing data from 30 individuals (10 patients and their unaffected parents). A filtering pipeline resulted in a first list of on average 3.8 candidate genes per patient. None of the known genes associated with hypopituitarism were found using this approach. A comparison with cDNA library data from embryonic mouse Rathke’s pouches showed that several of these candidate genes, including SLC20A1 and SLC15A4, were expressed in Rathke’s pouch tissue during early stages of pituitary development. Bearing in mind the limitations of cDNA library construction and the possible differences between human and murine pituitary organogenesis, the absence of a gene in these databases does not necessarily mean that it is not expressed in the pituitary at this particular developmental stage. For this reason, the other genes still represent possible candidates for CPHD.
Taking advantage of the trio-based approach (patients and unaffected parents), de novo variants were of particular interest. Using the pituitary transcriptome filter, four genes remained as candidates, inherited de novo. Given the oligogenic nature of CPHD and low recurrence rate of mutations among CPHD genes, we selected and sequenced SLC20A1 and SLC15A4 in additional 200 patients. These large-scale resequencing studies identified another likely pathogenic variant in SLC20A1 and one further likely pathogenic variant in SLC15A4.
SLC20A1 and SLC15A4 belong to the SLC group of membrane oligopeptide transporter superfamily.31 Most members, including SLC20A1 and SLC15A4, are located in the cell membrane. The SLC20 family transport proteins function as sodium phosphate cotransporters across the plasma membrane. SLC20A1 is expressed ubiquitously in all tissues and although generally considered housekeeping transport proteins, the discovery of tissue-specific activity, regulatory pathways, and gene-related pathophysiologies is redefining their importance.32 A Slc20a1 knockout mouse is embryonically lethal.33 We have identified two SLC20A1 variants, L89S and L521F, with high CADD scores in two unrelated individuals (Table 2). The variants reside in exon 1 and exon 7 in positions that are highly conserved between vertebrates; L521F was even conserved down to Drosophila.
Members of the SLC15A4 family are using proton motive force for uphill transport of free histidine and certain di- and tripeptides from the endosome to the cytoplasm.27 They are predicted to contain 12 transmembrane domains with N- and C-termini facing the cytosol mediating the uptake and reabsorption of protein digestion products into the intestine and kidney, and function in neuropeptide homeostasis in the brain.34 Unlike other members of the SLC15 family not much is known about the function of SLC15A4 in the brain.35,36 In the adult rat brain, Slc15a4 has been shown to be widely distributed and it can be detected in the hippocampus, cerebellum, pontine nucleus, cerebral cortex, brain stem, thalamus, and hypothalamus in neuronal and nonneuronal cells. It has been suggested that Slc15a4 functions in removal of degraded neuropeptides, e.g., neuromodulators from the synaptic cleft, as well as controls the uptake of oligopeptides to regulate the cellular metabolism in the central nervous system.36 A knockout mouse revealed no significant differences between wild type in serum chemistry, body weight, viability, and fertility.37 The variant P456L detected in one of our CPHD patients is located within a small loop between the transmembrane domains (TMD) 9 and 10 close to TMD10. The affected amino acid Pro is highly conserved within vertebrates as well as within the SLC15 family and was predicted as damaging by all prediction programs used in this study. The variant L84F resides in the TMD2 and was also predicted to affect protein function. How these variants affect protein structure and function in the brain linked to pituitary development will need to be elucidated in future studies. These studies may not be trivial, as only the combined analysis of all variants in one individual may lead to the deleterious consequences.
Large-scale genotyping and sequencing methods and access to large-scale population control exomes have only now allowed unbiased assessments of penetrance in population controls. In a second approach (Figure 1b), we raised the question whether likely damaging variants of known genes underlying CPHD have been transmitted from unaffected parents to affected children. When we went back to the initial set of 400 genes per patient and asked which likely damaging gene variants are shared between children and parents, we were surprised to detect that 3 of the 10 exome-sequenced patients also carried likely damaging variants in the GLI2 and LHX3 genes. Several variants of these genes have strong contextual support from preexisting genetic data.27,28 Heterozygous GLI2 mutations have been identified relatively frequently (17%) especially in patients with CPHD and an ectopic posterior pituitary lobe,38 while LHX3 mutations were found less frequently and all in homozygous state (Supplementary Table S4).27,28 We also identified a frameshift in the hypopituitary gene TCF7L1 (p.S15Kfs*41), transmitted from an unaffected mother to her son (RaKi_51), suggesting that this variant, possibly in concert with the de novo mutation in NOBOX, contributes to the CPHD phenotype in her son.29
In our study we focused on variants that are extremely rare or absent in the large contemporary multiethnic sequencing databases (e.g., gnomAD) (e.g., variants in SLC20A1 and SLC15A4), as we initially argued that a fully penetrant disease genotype should be no more common in the population than the disease that it causes. Likely damaging variants in these two candidate genes were also found in further 2 out of 200 patients, making it extremely likely that the variants are causative from a statistical perspective compared with individuals from the gnomAD server (0/55,607 individuals for the L89S variant of SLC20A1; 0/7506 individuals for the SLC15A4 P456L variant, and no data for the L521F variant of SLC20A1 and L84F variant of SLC15A4). We then provided additional evidence from variant prediction tools, pituitary transcriptomics, and IPA network analysis. Our analysis shows that the phenotype is derived from the combination of variants in more than one gene (oligogenic inheritance) and from a combination of both transmitted and de novo occurring variants in individual patients. Digenic inheritance in patients with CPHD did not come as a complete surprise and has been reported before.39
The novelty of our study is that in (at least) three of the exome-sequenced patients more than one gene contributed to the phenotype and that this oligogenic situation was a combination of rare de novo variants and transmitted variants. In patient RaKi_40, it may even be possible that, besides the likely damaging gene variant in SLC20A1 and the transmitted GLI2 variant, possibly damaging variants in the TET2 and AGAP1 genes (Supplementary Table S3) may contribute to the phenotype. All of these candidate genes are connected and activated via SOX2 (Figure 2).17,22 Mutations in SOX2 have a validated role in individuals with CPHD.26 Given the complexity of the issue, our exome data have opened new avenues and provided further evidence that CPHD is a genetically complex disorder with incomplete penetrance and variable expressivity.
Conclusion
In this report we demonstrate whole-exome sequencing as a successful approach to identify novel candidate genes underlying congenital hypopituitarism. Our study has provided new genetic risk loci for CPHD, which will contribute to the diagnostics of hypopituitarism as well as help to elucidate the molecular mechanisms underlying pituitary development. We have demonstrated that a combination of de novo and transmitted variants can occur in the same individuals and may contribute to the clinical phenotype in patients with CPHD. It is therefore possible that a combination of different gene variants is needed to lead to the full-blown phenotype. Locus-based sequencing strategies are not digging deep enough to reveal the overall pattern of alterations in this disorder and are not enough to evaluate diagnostic disease risk. Exome sequencing will remain the method of choice to provide sufficiently large data sets in disorders with incomplete penetrance and variable expressivity to allow a global genome-based view of all exonic genetic alterations.
References
Bancalari RE, Gregory LC, McCabe MJ, Dattani MT. Pituitary gland development: an update. Endocr Dev 2012;23:1–15.
Kelberman D, Rizzoti K, Lovell-Badge R, Robinson IC, Dattani MT. Genetic regulation of pituitary gland development in human and mouse. Endocr Rev. 2009;30:790–829.
Alatzoglou KS, Dattani MT. Genetic forms of hypopituitarism and their manifestation in the neonatal period. Early Hum Dev 2009;85:705–712.
Mehta A, Dattani MT. Developmental disorders of the hypothalamus and pituitary gland associated with congenital hypopituitarism. Best Pract Res Clin Endocrinol Metab 2008;22:191–206.
Pfaffle R, Klammt J. Pituitary transcription factors in the aetiology of combined pituitary hormone deficiency. Best Pract Res Clin Endocrinol Metab 2011;25:43–60.
Davis SW, Ellsworth BS, Perez Millan MI et al. Pituitary gland development and disease: from stem cell to hormone production. Curr Top Dev Biol 2013;106:1–47.
Phillips JA 3rd, Cogan JD. Genetic basis of endocrine disease. 6. Molecular basis of familial human growth hormone deficiency. J Clin Endocrinol Metab 1994;78:11–16.
Wu W, Cogan JD, Pfaffle RW et al. Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nat Genet 1998;18:147–149.
Mody S, Brown MR, Parks JS. The spectrum of hypopituitarism caused by PROP1 mutations. Best Pract Res Clin Endocrinol Metab 2002;16:421–431.
Cogan JD, Wu W, Phillips JA 3rd et al. The PROP1 2-base pair deletion is a common cause of combined pituitary hormone deficiency. J Clin Endocrinol Metab 1998;83:3346–3349.
Deladoey J, Fluck C, Buyukgebiz A et al. “Hot spot” in the PROP1 gene responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab 1999;84:1645–1650.
Lebl J, Vosahlo J, Pfaeffle RW et al. Auxological and endocrine phenotype in a population-based cohort of patients with PROP1 gene defects. Eur J Endocrinol 2005;153:389–396.
De Rienzo F, Mellone S, Bellone S et al. Frequency of genetic defects in combined pituitary hormone deficiency: a systematic review and analysis of a multicentre Italian cohort. Clin Endocrinol (Oxf) 2015;83:849–860.
Abecasis GR, Auton A, Brooks LD et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65.
Okazaki Y, Furuno M, Kasukawa T et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–573.
Willett RT, Greene LA. Gata2 is required for migration and differentiation of retinorecipient neurons in the superior colliculus. J Neurosci. 2011;31:4444–4455.
Adachi K, Nikaido I, Ohta H et al. Context-dependent wiring of Sox2 regulatory networks for self-renewal of embryonic and trophoblast stem cells. Mol Cell. 2013;52:380–392.
Cox JL, Wilder PJ, Gilmore JM, Wuebben EL, Washburn MP, Rizzino A. The SOX2-interactome in brain cancer cells identifies the requirement of MSI2 and USP9X for the growth of brain tumor cells. PLoS One. 2013;8:e62857.
Schepers G, Wilson M, Wilhelm D, Koopman P. SOX8 is expressed during testis differentiation in mice and synergizes with SF1 to activate the Amh promoter in vitro. J Biol Chem. 2003;278:28101–28108.
Fukami M, Wada Y, Okada M et al. Mastermind-like domain-containing 1 (MAMLD1 or CXorf6) transactivates the Hes3 promoter, augments testosterone production, and contains the SF1 target sequence. J Biol Chem. 2008;283:5525–5532.
Rasmussen KD, Jia G, Johansen JV et al. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 2015;29:910–922.
Whyte WA, Orlando DA, Hnisz D et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319.
Kallin EM, Rodriguez-Ubreva J, Christensen J et al. Tet2 facilitates the derepression of myeloid target genes during CEBPalpha-induced transdifferentiation of pre-B cells. Mol Cell. 2012;48:266–276.
Goldsmith S, Lovell-Badge R, Rizzoti K. SOX2 is sequentially required for progenitor proliferation and lineage specification in the developing pituitary. Development. 2016;143:2376–2388.
Charles MA, Saunders TL, Wood WM et al. Pituitary-specific Gata2 knockout: effects on gonadotrope and thyrotrope function. Mol Endocrinol 2006;20:1366–1377.
Kelberman D, Dattani MT. Role of transcription factors in midline central nervous system and pituitary defects. Endocr Dev 2009;14:67–82.
Arnhold IJ, Franca MM, Carvalho LR, Mendonca BB, Jorge AA. Role of GLI2 in hypopituitarism phenotype. J Mol Endocrinol 2015;54:R141–150.
Sobrier ML, Brachet C, Vie-Luton MP et al. Symptomatic heterozygotes and prenatal diagnoses in a nonconsanguineous family with syndromic combined pituitary hormone deficiency resulting from two novel LHX3 mutations. J Clin Endocrinol Metab 2012;97:E503–509.
Gaston-Massuet C, McCabe MJ, Scagliotti V et al. Transcription factor 7-like 1 is involved in hypothalamo-pituitary axis development in mice and humans. Proc Natl Acad Sci USA. 2016;113:E548–557.
Webb EA, AlMutair A, Kelberman D et al. ARNT2 mutation causes hypopituitarism, post-natal microcephaly, visual and renal anomalies. Brain 2013;136:3096–3105.
Colas C, Ung PM, Schlessinger A. SLC transporters: structure, function, and drug discovery. Medchemcomm. 2016;7:1069–1081.
Forster IC, Hernando N, Biber J, Murer H. Phosphate transporters of the SLC20 and SLC34 families. Mol Aspects Med. 2013;34:386–395.
Beck L, Leroy C, Beck-Cormier S et al. The phosphate transporter PiT1 (Slc20a1) revealed as a new essential gene for mouse liver development. PLoS One. 2010;5:e9148.
Smith DE, Clemencon B, Hediger MA. Proton-coupled oligopeptide transporter family SLC15: physiological, pharmacological and pathological implications. Mol Aspects Med. 2013;34:323–336.
Botka CW, Wittig TW, Graul RC et al. Human proton/oligopeptide transporter (POT) genes: identification of putative human genes using bioinformatics. AAPS PharmSci. 2000;2:E16.
Yamashita T, Shimada S, Guo W et al. Cloning and functional expression of a brain peptide/histidine transporter. J Biol Chem. 1997;272:10205–10211.
Wang XX, Hu Y, Keep RF, Toyama-Sorimachi N, Smith DE. A novel role for PHT1 in the disposition of l-histidine in brain: in vitro slice and in vivo pharmacokinetic studies in wildtype and Pht1 null mice. Biochem Pharmacol 2017;124:94–102.
Franca MM, Jorge AA, Carvalho LR et al. Relatively high frequency of non-synonymous GLI2 variants in patients with congenital hypopituitarism without holoprosencephaly. Clin Endocrinol (Oxf) 2013;78:551–557.
Sykiotis GP, Plummer L, Hughes VA et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci USA 2010;107:15140–15144.
Acknowledgments
We thank the patients and the families for their kind support and Rafiullah Rafiullah for help with the sequencing. The authors also thank the Genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org/about) and the groups that provided exome and genome variant data to this resource.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Simm, F., Griesbeck, A., Choukair, D. et al. Identification of SLC20A1 and SLC15A4 among other genes as potential risk factors for combined pituitary hormone deficiency. Genet Med 20, 728–736 (2018). https://doi.org/10.1038/gim.2017.165
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.165
This article is cited by
-
Oligopeptide/Histidine Transporter PHT1 and PHT2 — Function, Regulation, and Pathophysiological Implications Specifically in Immunoregulation
Pharmaceutical Research (2023)
-
Refinement of evolutionary medicine predictions based on clinical evidence for the manifestations of Mendelian diseases
Scientific Reports (2019)

{kind=link}