
Abstract
Purpose
Disorders or differences of sex development (DSDs) are rare congenital conditions characterized by atypical sex development. Despite advances in genomic technologies, the molecular cause remains unknown in 50% of cases.
Methods
Homozygosity mapping and whole-exome sequencing revealed an ESR2 variant in an individual with syndromic 46,XY DSD. Additional cases with 46,XY DSD underwent whole-exome sequencing and targeted next-generation sequencing of ESR2. Functional characterization of the identified variants included luciferase assays and protein structure analysis. Gonadal ESR2 expression was assessed in human embryonic data sets and immunostaining of estrogen receptor-β (ER-β) was performed in an 8-week-old human male embryo.
Results
We identified a homozygous ESR2 variant, c.541_543del p.(Asn181del), located in the highly conserved DNA-binding domain of ER-β, in an individual with syndromic 46,XY DSD. Two additional heterozygous missense variants, c.251G>T p.(Gly84Val) and c.1277T>G p.(Leu426Arg), located in the N-terminus and the ligand-binding domain of ER-β, were found in unrelated, nonsyndromic 46,XY DSD cases. Significantly increased transcriptional activation and an impact on protein conformation were shown for the p.(Asn181del) and p.(Leu426Arg) variants. Testicular ESR2 expression was previously documented and ER-β immunostaining was positive in the developing intestine and eyes.
Conclusion
Our study supports a role for ESR2 as a novel candidate gene for 46,XY DSD.
Similar content being viewed by others
Introduction
In humans, gonadal fate is determined by the presence or absence of sex-specific gene expression. In the bipotential stage of development, transcription factors such as Wilms’ tumor 1, nuclear receptor subfamily 5 group A member 1 (NR5A1) and GATA-binding protein 4 (GATA4) lead to upregulation of the sex-determining region Y gene (SRY) in the male embryo, thereby initiating the male developmental signaling cascade, with the expression of other male-specific genes such as SRY-box 9 (SOX9).1 In addition, several reports indicate an important role for the mitogen-activated protein kinase (MAPK) pathway in the early phases of (male) sex determination.2,3,4 In female embryos lacking the Y chromosome, female-specific genes like R-spondin 1 and forkhead box L2 (FOXL2) are expressed among others.1,5 Animal studies have revealed multiple and long-lasting interactions between these male and female pathways; for instance, the loss of Foxl2 or Dmrt1 in murine adult granulosa and Sertoli cells, respectively, results in trans-differentiation to an opposite cell fate.6,7
Subtle imbalances of this strictly regulated sex-specific gene network can lead to disorders or differences of sex development (DSD).1 Currently, mutations in genes known to be involved in sex development explain the molecular pathogenesis in only a minority of 46,XY DSD cases.1,7 Next-generation-sequencing-based approaches such as whole-exome sequencing improve the identification of causal genetic defects, and the discovery of new genes and signaling pathways implicated in sex development.8,9 Elucidating the underlying causes of DSD can facilitate sex assignment and lead to improved management, a better prognosis and a more accurate evaluation of gonadal function and gonadal germ cell cancer risk.10
So far, mutations in genes encoding nuclear receptors, such as NR5A1 (also known as the steroidogenic factor 1 gene, or SF1)11 and the androgen receptor NR3C4 gene12 have been implicated in DSD pathogenesis as well as the duplication of the nuclear receptor subfamily 0 group B member 1 gene (NR0B1, DAX1).13 Here, we identified biallelic and monoallelic estrogen receptor 2 (ESR2) variants in one syndromic and two nonsyndromic 46,XY DSD cases, respectively, putting forward ESR2 as a novel disease gene for 46,XY DSD and expanding the spectrum of nuclear receptors implicated in 46,XY DSD etiology.
Materials and methods
Study subjects
Laboratory and clinical investigations of cases 1–3 are summarized in Table 1. Cases and/or parents provided written informed consent for the study. Detailed case reports can be found in Supplementary Data S1–S3 online.
Genetic studies
Homozygosity mapping was performed by genome-wide single-nucleotide polymorphism (SNP) genotyping in case 1 and in two unaffected siblings using the HumanCytoSNP-12 BeadChip platform (Illumina, San Diego, CA). Plink software, integrated in ViVar, identified homozygous regions of >1 Mb, which were ranked according to their length and the number of SNPs, as described.14 Only homozygous regions unique to the index case were selected. The presence of relevant known DSD genes was excluded using BioMart filtering. Next, different gene prioritization tools (Endeavour, GeneDistiller, GeneWanderer, PosMed, SUSPECTS, and ToppGene) were used to identify candidate genes in the selected regions.15 For this purpose, known DSD genes were used as training genes (Supplementary Data and Supplementary Table S1 online).
Sanger sequencing and targeted next-generation sequencing
Primers were developed for polymerase chain reaction amplification and sequencing of all coding exons, intron–exon boundaries and untranslated regions of ESR2 (Supplementary Data and Supplementary Table S2 online). Sanger sequencing (cases 1 and 2) was performed using an ABI 3730XL DNA Analyzer (Applied Biosystems, Carlsbad, CA) with the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) followed by data analysis with SeqScape Software v2.5 (Life Technologies, Europe, Ghent, Belgium). Targeted next-generation sequencing of 63 DSD disease and candidate genes including ESR2 was performed (case 3) with a MiSeq System instrument. Details of the target enrichment, variant annotation and filtering can be found in Supplementary Data S4 online.
Segregation analysis for the ESR2 variant found in case 1 was performed in the available family members. Apart from the mother, no family members were available for further analysis in case 2. For case 3, no parental DNA was available for segregation analysis. Variant classification was performed following the American College of Medical Genetics and Genomics (ACMG) guidelines of 2015.16
Whole-exome sequencing was performed to exclude the presence of other likely pathogenic variants. Exome enrichment was performed with the Nextera Rapid Capture Exome kit (Illumina), followed by paired-end sequencing on HiSeq 2000 (2 × 100 cycles) (Illumina). Reads were mapped against the human reference genome sequence (National Center for Biotechnology Information, GRCh37/hg19) with the CLC Genomics Workbench v6.4 (Qiagen, Venlo, The Netherlands) followed by postmapping duplicate read removal, coverage analysis, and quality-based variant calling. More thorough variant annotation was executed using Alamut-HT v1.1.5 software (Interactive Biosoftware, Rouen, France) and a complementary literature search after extensive screening (Supplementary Table S3–S5 online).
Targeted NR5A1 testing
In cases 1 and 2, targeted next-generation sequencing of the coding region of NR5A1 was performed using a previously described workflow,17 and copy-number variant analysis was performed using multiplex ligation-dependent probe amplification (SALSA MLPA P185 Intersex probemix; MRC-Holland, Amsterdam, The Netherlands). In case 3 NR5A1 sequence variants and copy-number variants were excluded by the aforementioned targeted next-generation sequencing panel analysis.
Structural analysis of the estrogen receptor-β (ER-β) variants
Models for the wild-type ER-β DNA-binding domain (DBD) were built using the standard homology modeling procedure in YASARA Structure (http://yasara.org/products). We used the structure of the ER-β bound to a DNA response element (pdb code 1HCQ) as a template.18 We built models for dimers of the wild-type DBD with and without DNA. A model for the p.(Asn181del) (N181del) mutation (found in case 1) was built in YASARA Structure based on the wild-type model, by deleting residue N181 and introducing a new peptide bond between residues 180 and 182, followed by energy minimization (the em_runclean protocol in YASARA Structure). The homodimer models for the wild-type and N181del mutant DBDs were further subjected to 500 ps of molecular dynamics with the YASARA2 forcefield, followed by energy minimization using the md_refine procedure.19
Using FoldX and the estrogen-bound ER-β ligand-binding domain (LBD) structure with pdb code 3OLL, we calculated an in silico estimate of the Gibbs energy of protein folding for the mutant and wild-type LBD using the RepairPDB and BuildModel commands.20,21 For each mutant, we report the average ΔΔG for the folding of mutant versus wild-type of 20 BuildModel calculations. The 3OLL protein structure was visualized in UCSF Chimera.22 The effect of the p.(Leu426Arg) (L426R) mutation (found in case 3) was visualized by replacing L426 with the best-fitting arginine rotamer in the Dunbrack backbone-dependent rotamer library.
ESR2 expression analysis in human embryonic tissue
Assessment of human embryonic expression data sets
ESR2 expression in the testis was assessed in RNA sequencing data generated by Gerrard et al.23 in human embryos of Carnegie stages 18 and 22 and in array expression data generated by del Valle et al.24 in embryonic gonadal and adrenal tissues.
Immunohistochemistry
Tissue slides (4 μm) were prepared from a formalin-fixed, paraffin-embedded 8-week-old male fetus obtained after spontaneous abortion. Before immunohistochemistry, a heat-induced antigen retrieval step (Tris-EGTA, pH 9.00) was included. We used a standard ABC protocol for the detection of ER-β. After blocking endogenous peroxidase and biotin (Avidin/Biotin Blocking Kit SP-2001) and overnight incubation with mouse anti-ER-β (dilution 1/50; Dako M7292), detection was performed with a biotinylated rabbit antimouse antibody (dilution 1/150, 30 min; Dako E413) followed by 30 min of incubation with Avidin–Biotin–HRP-labeled complex (VECTASTAIN Elite ABC HRP Kit PK-6100). Visualization was done with DAB/H2O2 and hematoxylin counterstaining.
Luciferase assays
Plasmid construction
Plasmids containing the ESR2 variants were constructed with site-directed mutagenesis starting from a pcDNA–flag–ER-β plasmid (35562; Addgene, Cambridge, MA). Mutagenesis primers were designed using the NEBaseChanger software (http://nebasechanger.neb.com/). After amplification, the plasmids were digested with DpnI to remove nonmutated DNA. After verifying that mutations had correctly been inserted via sequence analysis, mutated plasmids were transformed in chemically competent Escherichia coli (DH10b cells) and subsequently isolated with the NucleoBond Xtra Midi/Maxi kit (Macherey-Nagel, Düren, Germany).
Cell culture and luciferase assays
HEK293T cells were cultured in Dulbecco’s minimal essential medium without phenol red, supplemented with 10% charcoal-stripped fetal bovine serum (Life Technologies, Europe, Ghent, Belgium) and 10 μg/ml gentamycin at 37 °C and 8% CO2. A total of 300,000 cells per well were plated in 6-well plates. Cells were transfected with a total of 2 μg of DNA using a standard calcium phosphate transfection method. After 24 h, cells were washed with phosphate buffered saline and subsequently stimulated with 2,3-bis(4-hydroxyphenyl)propionitrile (DPN) to a final concentration of 10 nM. Following lysis with 1 × cell culture lysis reagent (25 mM Tris-phosphate pH 7.8, 2 mM dithiothreitol, 2 mM 1,2-cyclohexylenedinitrilotetraacetic acid, 10% glycerol, and 1% Triton X-100), luciferase activity was measured 24 h after stimulation on a TopCount luminometer (Canberra–Packard, Meriden, CT). To normalize for differences in transfection efficiency, a plasmid constitutively expressing β-galactosidase was cotransfected in all experiments (Tropix Galacto-Star system; Applied Biosystems, Foster City, CA). Each transfection was replicated in at least three independent experiments. Differences in expression between wild-type and mutated ER-β were demonstrated for variants 1 and 2 using a control western blot (data not shown).
Results
Identification of a homozygous ESR2 variant in case 1 with syndromic 46,XY DSD
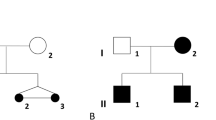
Given the possible parental consanguinity, we performed homozygosity mapping to identify runs of homozygosity containing the putative underlying genetic cause of the syndromic 46,XY DSD phenotype in case 1. Twenty-one homozygous regions larger than 1 Mb were identified (Supplementary Data and Supplementary Table S6 online). The largest one—with a length of 20 Mb—is located on chromosome 14 and contains 465 genes, none of which are known DSD genes. The gene encoding 5α-reductase, SRD5A2, was present in another small homozygous region (3.6 Mb); however, no potentially pathogenic variants were found in its coding region. Variant prioritization ranked ESR2 as the top candidate gene (Supplementary Data and Supplementary Table S7 online). Sequencing of ESR2 revealed a homozygous 3-bp deletion in exon 9, c.541_543del, leading to an in-frame deletion, p.(Asn181del) (Figure 1a). This deletion affects a highly conserved amino acid located in the DBD (Figure 1a). The deletion was found to be absent in 92 tested ethnically matched control chromosomes and 320 other tested control chromosomes. This variant is reported at very low frequencies in publicly available databases (the Exome Sequencing Project, the Exome Aggregation Consortium 0.3.1, and gnomAD); however, no homozygotes have previously been identified (variant details in Supplementary Data and Supplementary Table S8 online). Segregation analysis showed that both parents (Figure 1b; I:1 and I:2) and one healthy 46,XX sibling (Figure 1b; II:2) are heterozygous carriers of the deletion, while another 46,XX sibling (Figure 1b; II:3) is homozygous for the wild-type allele. Variant classification following the ACMG guidelines categorizes p.(Asn181del) as likely pathogenic (class 4) (Table 2). Whole-exome sequencing in the index case and the parents made the presence of other potentially pathogenic coding variants very unlikely. An overview of the variant filtering, performed as previously described,14 is given in Supplementary Tables S3 and S4 online.

(a) Schematic overview of the ER-β protein with detailed representation of the DNA-binding domain. The light gray circles represent P-box amino acids, the dark gray circles represent the dimerization interface and the red circle represents the deleted amino acid in case 1. (b–d) Left: protein alignment; middle: electropherogram; and right: pedigree, for the deleted amino acid Asn181 (N181) found in case 1 conserved up to Tetraodon and in human ER-α (b); the affected amino acid in case 2, Gly84 (G84) conserved up to the fruit fly (c); and the affected amino acid in case 3, Leu426 (L426) conserved up to Tetraodon (d). In (b–d), the affected individual is represented by a black symbol. M, mutation; NA, not available.
Identification of two heterozygous ESR2 missense variants in case 2 and 3 with nonsyndromic 46,XY DSD
Targeted resequencing of the ESR2 coding region in a Belgian 46,XY DSD cohort (n = 73) and a Brazilian 46,XY DSD cohort (n = 40) revealed two heterozygous missense variants in two unrelated cases with nonsyndromic 46,XY DSD. In case 2, a heterozygous ESR2 variant c.251G>T was found in exon 2, leading to a missense change p.(Gly84Val) affecting a highly conserved amino acid (up to fruit fly) located in the N-terminal domain, which contains the first activation function of the receptor (Figure 1a). The physicochemical difference between Gly and Val is moderate (a Grantham distance of 109). Several prediction programs (SIFT, PolyPhen-2, and MutationTaster) suggest a deleterious effect on protein function. The variant is absent in 412 tested control chromosomes and in genomic databases such as dbSNP, the Exome Sequencing Project, and the Exome Aggregation Consortium. In gnomAD, it occurs in 0.000396% (no homozygotes) (Table 2). Following the ACMG guidelines, p.(Gly84Val) could be scored as likely pathogenic (class 4) (Table 2). Whole-exome sequencing did not reveal any other potentially pathogenic variants in case 2. An overview of the different filtering steps and variants is given in Supplementary Tables S3 and S5 online. In case 3, a heterozygous variant—c.1277T>G in exon 8 of ESR2—was found, leading to the missense change p.(Leu426Arg) in the LBD and affecting a highly conserved amino acid (up to Tetraodon). The Grantham distance between Leu and Arg is moderate (i.e., 102) and several prediction programs show a potential deleterious effect. This variant is not present in the consulted genomic databases (dbSNP, the Exome Sequencing Project, the Exome Aggregation Consortium, and gnomAD), nor in 214 Brazilian exomes (Table 2). Following the ACMG guidelines, p.(Gly84Val) could be classified as a variant of unknown significance (class 3) (Table 2). Whole-exome sequencing did not reveal any other potentially pathogenic variants in case 3. Figure 1 and Table 2 summarize the location and characteristics of the identified ESR2 variants.
Structural analysis of the ER-β variants
The ER-β DBD binds to DNA as a homodimer. We built homology models for the wild-type and p.(Asn181del) mutant homodimer. Residue Asn181 is part of a longer extended loop that connects the main DNA-binding helix to a zinc coordination site, which is part of the homodimer interface. The Asn181 deletion shortens this connecting loop and causes a 15° tilt of the main DNA-binding helix (Figure 2a). Molecular dynamics simulation, followed by energy minimization, suggests that this slightly affects the orientation between the two DBDs in the homodimer, with an increased number of contacts between the two domains (Figure 2a). It is therefore very likely that the Asn181 deletion affects homodimerization and/or DNA binding of the ER-β.

(a) Effect of the Asn181 deletion on a dimer of the ER-β DNA-binding domain. A model for a dimer of both the wild-type (blue) and Asn181del mutant (red) DNA-binding domain is shown. Monomer 1 of both dimers is superposed. The Asn181 (N181) deletion shortens a long loop between the DNA-binding helix and the zinc-binding site at the dimer interface. As a result, the DNA-binding helix tilts towards the dimer interface. The red arrow indicates an upward shift of the second monomer versus the first monomer in the mutant dimer. The molecules are rotated 90° compared with (b) and the DNA-binding helices are directed towards the viewer. (b) Model of the wild-type ER-β DNA-binding domain bound to DNA. The position of Asn181 is indicated. (c) Position of the Leu270 (L270) and Leu426 (L426) residues in the ER-β ligand-binding domain structure. Atoms of L270 and L426 are shown as gray spheres. (d) L426 is buried in a hydrophobic environment in the ligand-binding domain structure. (e) When L426 is mutated to the best-fitting Arg rotamer in UCSF Chimera, the Arg side chain clashes with residues D326 and M410 (red lines).
There was no template available for remodeling of the p.(Gly84Val) variant, and no homology models could be obtained. To assess the possible influence of the p.(Leu426Arg) missense variant on the ER-β protein structure, we made in silico calculations of ΔΔG with FoldX and tried to model the best rotamer position for each variant with UCSF Chimera. The p.(Leu426Arg) variant is located in the LBD, which was previously characterized by X-ray crystallography. Leu426 is a buried residue of helix 9 (Figure 2a). FoldX energy calculations using the BuildModel command indicate that the p.(Leu426Arg) mutation affects the folding energy of the protein and has a destabilizing effect (in silico ΔΔG = 2.3 kcal/mol). Leu426 fits in a hydrophobic pocket and engages in interactions with Leu322, Leu330, Leu406, and Met410 (Figure 2b). Modeling the best-fitting Arg rotamer at the position of Leu426 in UCSF Chimera shows that this mutation abolishes most of these hydrophobic interactions, while even the best-fitting rotamer of the Arg side chain introduces strong steric clashes in the absence of drastic energy minimization (green lines) (Figure 2c). The p.(Leu426Arg) variant is likely to affect the stability or folding of the LBD domain. Folding of the p.(Leu426Arg) variant estrogen-bound LBD domain into a wild type-like structure would at least require drastic structural changes in the environment of residue 426, which could hamper LBD dimerization, as helix 9 and the loop between helix 8 and 9 (which interacts with Leu426 via Met410) are both part of the symmetrical LBD dimer interface.
Expression studies in human embryos
In RNA sequencing data generated by Gerrard et al.23 in embryos of Carnegie stages 18 and 22 (7 and 8 weeks; i.e., around the time of sex determination), ESR2 expression could be observed in testicular tissues. In array expression data generated by del Valle et al.24 in a genomic atlas of human adrenal and gonadal development, early expression of ESR2 could be confirmed. In a human male embryo with a gestational age of 8 weeks, ER-β immunostaining was positive in the developing eye, especially in the corneal epithelium and endothelium, primitive lens neurons, primitive retinal cells and retinal pigment epithelium, mucosa of the developing intestine, and neuronal system (specifically the primitive neurons of the germinal matrix). ER-β staining was negative in the developing testis at this stage of development (Supplementary Data and Supplementary Figure S1 online).
Transcriptional activation assays
The impact of the identified variants on transcriptional activation was assessed using luciferase assays with TK-3xERE-promoter constructs. The p.(Asn181del) variant resulted in significantly increased transcriptional activation after stimulation with the ER-β-specific ligand, DPN (P < 0.001). For the two missense variants, p.(Gly84Val) and p.(Leu426Arg), no significant alterations of ligand-dependent transcriptional activation were observed (Figure 3). Interestingly, the p.(Leu426Arg) variant shows a higher transcriptional activation compared with wild-type ER-β when the cells are not stimulated with DPN (P = 0.0014).

Results are shown as the mean ± SEM for relative luciferase intensity units (R.L.U.). The analysis involved the fitting of a hierarchical generalized linear model to the R.L.U. data, as implemented in Genstat v18, with transfection and stimulation as fixed terms (Poisson distribution; log link) and replicates (n = 4 or higher) nested within experimenter as additional random terms (gamma distribution; log link). **P < 0.01. The difference between WT and Asn181del is significant at the 0.001 level. For variants 1 and 2, the experiment was repeated 11 times respectively with three to six technical replicates each time; for variant 3, the experiment was repeated seven times with six replicates each time. DMSO, dimethylsulfoxide; DPN, 2,3-bis(4-hydroxyphenyl)propionitrile.
Discussion
The identification of likely pathogenic variants in ESR2 in unrelated individuals with 46,XY gonadal dysgenesis suggests an unanticipated role for ER-β in early gonadal development. Previously, polymorphic ESR2 variants have been associated with milder variations in sexual development such as hypospadias and micropenis,25,26,27,28,29 and a heterozygous ESR2 variant c.1295C>A leading to a missense change p.(Ala432Asp) in the LBD of ER-β was recently reported in a girl with primary amenorrhea.30
Here, we identified a biallelic in-frame deletion in a girl with syndromic 46,XY DSD and two monoallelic missense variants in two unrelated cases with nonsyndromic 46,XY DSD. The data of case 1 are suggestive of absent gonadal development, while the data of cases 2 and 3 are compatible with partial and complete gonadal dysgenesis. A combination of different parameters including population frequencies and in silico predictions support a likely pathogenic classification for two of the three variants found (Table 2). Structural analyses support a deleterious effect of the p.(Asn181del) and p.(Leu426Arg) variants on protein conformation, but are not available for the p.(Gly84Val) variant due to the absence of a suitable model for this region. A significant increase in transcriptional activation was shown for the p.(Asn181del) and p.(Leu426Arg) variants and resulted in increased activity in the absence of an ER-β-specific ligand.
In recently published human embryonic gonadal expression databases, ESR2 expression was found in early stages of gonadal development. In an 8-week-old male human embryo, ER-β immunostaining was shown in the developing intestine, eye, and neuronal systems, potentially reflecting the extragonadal involvement in case 1. However, no testicular immunostaining was observed at this developmental time point. Given the severity of the observed gonadal phenotypes, it cannot be excluded that ER-β plays a role even earlier during development.
An interesting finding in this respect is the fact that nonclassical estrogen signaling has been shown previously to interact with the MAPK pathway31—a signaling pathway involved in the earliest stages of gonadal development—although the operating mechanisms are different in humans and mice. In humans, MAPK signaling is essential for tilting the gene expression balance towards SOX9 expression; gain-of-function mutations of MAP3K1 lead to higher phosphorylation of downstream targets, causing decreased expression of SOX9 and increased expression of female-specific genes like β-catenin.2,31,32 In mice, the Mapk pathway acts through direct upregulation of Sry; homozygous loss-of-function variants of Map3k4 are reflected in a male-to-female sex reversal phenotype.3,33
Nuclear receptor family members, to which the ERs belong, are known to exert their functions through different action mechanisms. The classical genomic pathway involves ligand binding and receptor dimerization, followed by nuclear translocation and activation or repression of target gene expression.34,35 Estrogen binds on both ER-α and ER-β receptors; however, binding results in partially overlapping but also differential transcriptional effects.36,37 Alternative action mechanisms involve interactions with other transcription factors, ligand-independent receptor activation, and activation of different signaling pathways. For instance, it has been shown that ER-β can activate specific MAPK pathways in human colon cancer cell lines. The scarce experimental data regarding these alternative pathways indicate that these processes are highly cell specific, making it hard to investigate them in a development gonadal context as the currently available cell lines do not mimic the embryonic cellular environment. We hypothesize that the ESR2 variants found here could lead to increased activation of the MAPK pathway via a nongenomic working mechanism, thereby shifting the expression balance from SOX9 to β-catenin signaling, and resulting in decreased SOX9 activation and failure of testicular development (Supplementary Data and Supplementary Figure S2 online). Experimental validation of this hypothesis is challenging given the cell-type specificity of these alternative pathways and the fact that no relevant cell line resembling the undifferentiated gonad is available. Furthermore, it remains complex to acquire an experimental setup that resembles the earliest phases of sex development as all available gonadal cell lines are differentiated.
A differential action mode of MAPK signaling in mice and humans can explain the absence of a sex reversal phenotype in ER-β knockout mouse models. Over the years, several ER-β KO mice have been generated. A first mouse knockout model was characterized by female sub- or infertility and normal reproductive capacity in males.38 This mouse was generated through insertion of a neomycin resistance gene into exon 3 of the Esr2 gene by homologous recombination in embryonic stem cells. Several transcripts were still being processed, however, leading to an incomplete knockout. Moreover, ER-β knockout mice originating from the same initial ER-β mutant but raised and examined in different laboratories presented with a range of different phenotypes. A new ER-β knockout mouse used Cre/LoxP-mediated excision of the third exon of Esr2, resulting in complete absence of any Esr2 transcript. These mice show infertility in both sexes, but no sex reversal. A similar situation has been reported for another nuclear receptor, NR0B1 (DAX1), for which duplications lead to male-to-female sex reversal in humans, but delayed testis development in mice.39
Differences in zygosity have been shown for the ESR2 variants found here, and may relate to the nonsyndromic phenotype in the two monoallelic cases compared with the syndromic phenotype in the biallelic case. Moreover, a different working mechanism cannot be excluded for the monoallelic missense variants found, although they may all act via the same dysregulated MAPK pathway. Differences in zygosity and mutation-dependent action mechanisms have been described for other nuclear receptors, such as NR2E3, implicated in both autosomal dominant and recessive retinal dystrophies with clinically distinct phenotypes.40
Conclusion
We identified biallellic and monoallelic variants in ESR2 in patients with syndromic and nonsyndromic 46,XY DSD, respectively, putting forward ESR2 as a novel candidate gene for 46,XY DSD etiology. It is interesting to speculate about an unanticipated role for ER-β in early gonadal development; i.e., the development and stabilization of the bipotential gonad and/or its differentiation into a functional testis.
References
Ono M, Harley VR. Disorders of sex development: new genes, new concepts. Nat Rev Endocrinol 2013;9:79–91.
Pearlman A, Loke J, Le Caignec C et al. Mutations in MAP3K1 cause 46,XY disorders of sex development and implicate a common signal transduction pathway in human testis determination. Am J Hum Genet 2010;87:898–904.
Bogani D, Siggers P, Brixey R et al. Loss of mitogen-activated protein kinase kinase kinase 4 (MAP3K4) reveals a requirement for MAPK signalling in mouse sex determination. PLoS Biol 2009;7:e1000196.
Warr N, Bogani D, Siggers P et al. Minor abnormalities of testis development in mice lacking the gene encoding the MAPK signalling component, MAP3K1. PLoS ONE 2011;6:e19572.
Uhlenhaut NH, Jakob S, Anlag K et al. Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell 2009;139:1130–1142.
Matson CK, Murphy MW, Sarver AL, Griswold MD, Bardwell VJ, Zarkower D. DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature 2011;9:1–5.
Hughes IA, Houk C, Ahmed SF, Lee PA. Consensus statement on management of intersex disorders. Arch Dis Child 2006: 554–563.
Bashamboo A, Ledig S, Wieacker P, Achermann J, McElreavey K. New technologies for the identification of novel genetic markers of disorders of sex development (DSD). Sex Dev 2010;15:213–224.
Baxter RM, Arboleda VA, Lee H et al. Exome sequencing for the diagnosis of 46, XY disorders of sex development. J Clin Endocrinol Metab 2015;100:E333–E344.
Cools M, Wolffenbuttel KP, Drop SLS, Oosterhuis JW, Looijenga LHJ. Gonadal development and tumor formation at the crossroads of male and female sex determination. Sex Dev 2011;5:167–180.
Ferraz-de-Souza B, Lin L, Achermann JC. Steroidogenic factor-1 (SF-1, NR5A1) and human disease. Mol Cell Endocrinol 2011;336:198–205.
Mongan N, Tadokoro-Cuccaro R, Bunch T, Hughes I. Androgen insensitivity syndrome. Best Pract Res Clin Endocrinol Metab 2015;29:569–580.
Orekhova AS, Rubtsov PM. DAX1, an unusual member of the nuclear receptor superfamily with diverse functions. Mol Biol 2015;49:65–76.
Coppieters F, Van Schil K, Bauwens M et al. Identity-by-descent-guided mutation analysis and exome sequencing in consanguineous families reveals unusual clinical and molecular findings in retinal dystrophy. Genet Med 2014;16:671–680.
Tranchevent L-C, Capdevila FB, Nitsch D, De Moor B, De Causmaecker P, Moreau Y. A guide to web tools to prioritize candidate genes. Brief Bioinform 2011;12:22–32.
De Leeneer K, Hellemans J, Steyaert W et al. Flexible, scalable, and efficient targeted resequencing on a benchtop sequencer for variant detection in clinical practice. Hum Mutat 2015;36:379–387.
Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424.
Schwabe JWR, Chapman L, Finch JT, Rhodes D. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell 1993;75:567–578.
Krieger E, Joo K, Lee J et al. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins 2009;77:114–122.
Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol 2002;320:369–387.
Möcklinghoff S, Rose R, Carraz M, Visser A, Ottmann C, Brunsveld L. Synthesis and crystal structure of a phosphorylated estrogen receptor ligand binding domain. Chembiochem 2010;11:2251–2254.
Pettersen EF, Goddard TD, Huang CC et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–1612.
Gerrard DT, Berry AA, Jennings RE, Hanley KP, Bobola N, Hanley NA. An integrative transcriptomic atlas of organogenesis in human embryos. eLlife 2016;5:1–16.
Del Valle I, Buonocore F, Duncan AJ et al. A genomic atlas of human adrenal and gonad development. Wellcome Open Res 2017;2:25.
Ban S, Sata F, Kurahashi N et al. Genetic polymorphisms of ESR1 and ESR2 that may influence estrogen activity and the risk of hypospadias. Hum Reprod 2008;23:1466–1471.
Beleza-Meireles A, Omrani D, Kockum I, Frisén L, Lagerstedt K, Nordenskjöld A. Polymorphisms of estrogen receptor β gene are associated with hypospadias. J Endocrinol Invest 2006;29:5–10.
Beleza-Meireles A, Kockum I, Lundberg F, Söderhäll C, Nordenskjöld A. Risk factors for hypospadias in the estrogen receptor 2 gene. J Clin Endocrinol Metab 2007;92:3712–3718.
Van der Zanden LFM, van Rooij IALM, Feitz WFJ, Franke B, Knoers NVAM, Roeleveld N. Aetiology of hypospadias: a systematic review of genes and environment. Hum Reprod Update 2012;18:260–283.
Van der Zanden LF, van Rooij IA, Feitz WF, Franke B, Knoers NV, Roeleveld N. Genetics of hypospadias: are single-nucleotide polymorphisms in SRD5A2 ESR1 ESR2, and ATF3 really associated with the malformation? J Clin Endocrinol Metab 2010;95:2384–2390.
Asadi M, Ghafouri-Fard S, Zare-Abdollahi D, Ebrahim-Habibi A, Matin N. Estrogen receptor mutation in a girl with primary amenorrhea. Clin Genet 2013;83:497–498.
Drummond AE, Fuller PJ. The importance of ERbeta signalling in the ovary. J Endocrinol 2010;205:15–23.
Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics 2006;7:497–508.
Loke J, Pearlman A, Radi O et al. Mutations in MAP3K1 tilt the balance from SOX9/FGF9 to WNT/β-catenin signaling. Hum Mol Genet 2014;23:1073–1083.
Ostrer H. Disorders of sex development (DSDs): an update. J Clin Endocrinol Metab 2014;99:1503–1509.
Pettersson K, Gustafsson JA. Role of estrogen receptor beta in estrogen action. Annu Rev Physiol 2001;64:65–92.
Segars JH, Driggers PH. Estrogen action and cytoplasmic signaling cascades. Part I: membrane-associated signaling complexes. Trends Endocrinol Metab 2002;13:349–354.
Driggers PH, Segars JH. Estrogen action and cytoplasmic signaling pathways. Part II: the role of growth factors and phosphorylation in estrogen signaling. Trends Endocrinol Metab 2002;13:422–427.
Krege JH, Hodgin JB, Couse JF et al. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci USA 1998;95:15677–15682.
Swain A, Narvaez V, Burgoyne P, Camerino G, Lovell-badge R. Dax1 antagonizes Sry action in mammalian sex determination. Nature 1998;391:761–767.
Coppieters F, Leroy BP, Beysen D et al. Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa. Am J Hum Genet 2007;81:147–157.
This work was supported by grants from the Research Foundation—Flanders (FWO) (Bilateral Scientific Cooperation Brazil NXT-DSDG0D6713N to E.D.B. and M.C.), the Ghent University Special Research Fund (BOF15/GOA/011 to E.D.B. and a BOF Starting Grant to M.C.), and Belspo IAP project P7/43 (Belgian Medical Genomics Initiative to E.D.B). E.D.B. and M.C. are senior clinical investigators of the Research Foundation—Flanders. H.V. is a postdoctoral fellow of the Research Foundation—Flanders. We are most grateful to the family members who participated in this study.
ESR2 STUDY GROUP
Zeynep Atay (Pediatric Endocrinology and Diabetes, Marmara University, Turkey); Abdullah Bereket (Pediatric Endocrinology and Diabetes, Marmara University, Turkey); Ronald R de Krijger (Reinier de Graaf Hospital, Delft, The Netherlands); Katleen de Preter (Center for Medical Genetics, Ghent University and Ghent University Hospital, Belgium); Sorahia Domenice (Disciplina de Endocrinologia, Laboratorio de Hormonios e Genetica Molecular LIM/42, Unidade de Adrenal, Disciplina de Endocrinologia e Metabologia, Hospital das Clínicas, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil); Serap Turan (Pediatric Endocrinology and Diabetes, Marmara University, Turkey).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflict of interest.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Baetens, D., Güran, T., Mendonca, B. et al. Biallelic and monoallelic ESR2 variants associated with 46,XY disorders of sex development. Genet Med 20, 717–727 (2018). https://doi.org/10.1038/gim.2017.163
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.163
This article is cited by
-
Genetic control of typical and atypical sex development
Nature Reviews Urology (2023)
-
Molecular mechanisms underlying AMH elevation in hyperoestrogenic states in males
Scientific Reports (2020)
-
Next-generation sequencing reveals genetic landscape in 46, XY disorders of sexual development patients with variable phenotypes
Human Genetics (2018)
