
Abstract
Purpose
CaV3.2 signaling contributes to nociception, pruritus, gastrointestinal motility, anxiety, and blood pressure homeostasis. This calcium channel, encoded by CACNA1H, overlaps the human tryptase locus, wherein increased TPSAB1 copy number causes hereditary α-tryptasemia. Germ-line CACNA1H variants may contribute to the variable expressivity observed with this genetic trait.
Methods
Tryptase-encoding sequences at TPSAB1 and TPSB2, and TPSG1 and CACNA1H variants were genotyped in 46 families with hereditary α-tryptasemia syndrome. Electrophysiology was performed on tsA201 HEK cells transfected with wild-type or variant CACNA1H constructs. Effects on clinical phenotypes were interrogated in families with TPSAB1 duplications and in volunteers from the ClinSeq cohort.
Results
Three nonsynonymous variants in CACNA1H (rs3751664, rs58124832, and rs72552056) cosegregated with TPSAB1 duplications in 32/46 families and were confirmed to be in linkage disequilibrium (LD). In vitro, variant CaV3.2 had functional effects: reducing current densities, and altering inactivation and deactivation properties. No clinical differences were observed in association with the CACNA1H haplotype.
Conclusion
A previously unrecognized haplotype containing three functional CACNA1H variants is relatively common among Caucasians, and is frequently coinherited on the same allele as additional TPSAB1 copies. The variant CACNA1H haplotype, which in vitro imparts partial gain of function, does not result in detectable phenotypic differences in the heterozygous state.
Similar content being viewed by others

Introduction
Increased α-tryptase encoding copy number at TPSAB1 is linked to elevated basal serum tryptase and is associated with multisystem complaints—a condition called hereditary α-tryptasemia syndrome.1 A gene-dose effect has been observed in this disorder, where a greater number of α-tryptase encoding copies on a single allele are associated with both increased basal serum tryptase level and prevalence of symptoms. However, the mechanism by which tryptase protein may contribute to clinical symptoms remains unclear. Owing to the structural complexity of the tryptase locus, germ-line sequence of this portion of the linkage region identified in hereditary α-tryptasemia, which includes the extra TPSAB1 copies, has not been fully ascertained. Given this, and the diffuse nature of clinical phenotypes observed, we hypothesized that coinherited variants may contribute to one or more of the clinical manifestations observed among affected individuals.
Within the linkage region (Chr16: 556,104–5,653,182; GRCh37/hg19), adjacent to the tryptase locus, exists the CACNA1H gene, which encodes the voltage-dependent, T-type, α1H subunit of the calcium channel, CaV3.2. While CACNA1H is a large and moderately mutable gene (Phred-scaled gene damage index = 11.351),2 functional variants in this T-type voltage-gated calcium channel would make a compelling candidate for many clinical phenotypes associated with TPSAB1 duplications. CaV3.2 signaling has been demonstrated as contributing to irritable bowel syndrome in a rat model of this disorder.3 Heightened anxiety, which can both contribute to and manifest as, symptoms associated with mast cell mediator release, has been reported in animal models with CACNA1H null alleles.4 While specific effects of this channel on autonomic nervous system function have not been reported, gain-of-function mutations have been identified in children with early onset hypertension and absence seizures.5,6 Non–mast cell–related, histamine-independent models of pruritus have also been shown to be dependent upon CaV3.2 expression.7 Finally, in addition to itch, a number of variants in voltage-gated calcium channels, including CACNG2 and CACNAD2D3, have been associated with chronic pain, and CaV3.2-dependent signaling has been shown to enhance pain responses in mice.8,9
We set out to characterize the frequency and effects of CACNA1H variants frequently coinherited with additional TPSAB1 copy, and determine how these affect channel function and clinical phenotypes both among individuals with hereditary α-tryptasemia syndrome and among unselected individuals.
Materials and methods
Subjects
Hereditary α-tryptasemia cohort
Informed consent was provided by all patients and their relatives on NIH IRB-approved research protocols designed to study mastocytosis (NCT00044122 and NCT00001756) and/or atopy (NCT01164241, NCT00852943, and NCT00557895). Family and personal medical histories were obtained, and physical examinations were performed as described.1
ClinSeq cohort
All study participants provided written informed consent and were actively enrolled in ClinSeq (NCT00410241), an IRB-approved genomic study conducted via the National Human Genome Research Institute.10 For inclusion in ClinSeq patients had to be: between the ages of 45 and 65 years, tobacco-smoke-free for at least 1 year, able to return frequently at their own expense to the NIH in Bethesda, MD, and have a primary care physician or have access to a community health center. Patients also could not have a nuclear family member concurrently enrolled in ClinSeq.
Genotyping and DNA sequence analysis
For the hereditary α-tryptasemia cohort, exome sequencing was performed on eight families using TruSeq (Illumina, San Diego, CA) capture kits and a custom analysis pipeline as described.11 Genome sequencing was performed as described12 on nine families (five of which previously had exome sequencing performed) using the HiSeq platform (Illumina) with the Burrows-Wheeler Aligner and Picard (http://broadinstitute.github.io/picard/) used for basic alignment and sequence quality control. For the ClinSeq cohort, exome sequencing was performed as described.13 The gene damage index for CACNA1H was obtained using the publicly available website http://pec630.rockefeller.edu:8080/GDI/.2 Linkage disequilibrium calculations were performed using Haploview (Broad Institute) as described.14 Recombination rate distribution in chromosome 16 near CACNA1H and TPSG1 was generated based on 1000 Genomes–phased OMNI data for CEU population (ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/working/20130507_omni_recombination_rates/CEU_omni_recombination_20130507.tar).
TPSAB1 and TPSB2 genotyping and CACNA1H haplotype determination
Allelic α- and β-encoding tryptase copy number at TPSAB1 was determined using digital droplet PCR (ddPCR) with use of custom primers and probes as described.1 TPSG1 and CACNA1H variant genotyping was performed using validated primer/probe sets for rs143120059 (C16107914), rs113856625 (C15283055), and rs72552056 (C99547390), rs3751664 (C25746770), rs58124832 (C90589833), respectively (ThermoFisher, Waltham, MA) by both RT-PCR and ddPCR; the results were validated by exome and genome sequence variant calls. Only individuals with all or none of the three CACNA1H variants were included in the phenotypic analysis.
ClinSeq individual selection and clinical phenotyping of all subjects
For clinical phenotypic analysis, haplotype carriers were defined as individuals with at least one allelic copy each of three different variants in CACNA1H passing quality filters, which were observed in strong linkage disequilibrium across the cohort: rs3751664 (NM_001005407:c.C2362T:p.(R788C)), rs58124832 (NM_001005407:c.G5594A:p.(R1865Q)), and rs72552056 (NM_001005407:c.C5995T:p.(R1999C)). Wild-type (WT) individuals were defined by homozygosity of the dominant allele at all three variant loci; these samples also met quality filters and had a minimum 10 × coverage depth at all three variant loci. Basal serum tryptase levels were measured in identified individuals, and the TPSAB1 copy number was determined among those with tryptase >8 ng/ml; those identified with increased TPSAB1 copy number were excluded from further evaluation.
A standardized questionnaire was administered to assess common clinical features associated with increased TPSAB1 copy number, seen in hereditary α-tryptasemia syndrome as described.1 Included in the survey were previously validated question sets from COMPASS 31 and Rome III.15,16 Patients were contacted by phone in random order by blinded interviewers, and the data were subsequently assigned to the appropriate genotype groups.
Site-directed mutagenesis
The variant CACNA1H construct (CACNA1HVAR) was created using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA) from a wild-type (WT) CACNA1H (CACNA1HWT) cDNA (transcript variant 2, NM_001005407.1) pcDNA3.1 vector (a kind gift from G.W. Zamponi, University of Calgary, Calgary, Alberta, Canada)). Mutation primers were designed using NEBaseChanger and obtained from UCDNA Services DNA/RNA Synthesis Laboratory (University of Calgary) as follows: R788C forward: GCGGCAAGCTtcgctGCATCGTGGA and reverse: TGAAGGTAACCCAGAGGCGGC; R1865Q forward: AAGGAGGCACaGGAGGATGCG and reverse: GTTGCTCTCCTCCAGGTG; R1999C forward: CTCGGGGCACcgcctGCTCCCCCAG and reverse: GGGACAGGGCGTGGAGGG. cDNAs were purified from transformed DH5α bacteria (MiniPrep kit; Qiagen, Hilden, Germany). Site-directed mutagenesis products and CACNA1HVAR were sequence verified (University Core DNA Services, University of Calgary).
Transfection and cell culture
tsA201 human embryonic kidney (HEK) cells were transiently transfected with 3 μg of plasmid and 0.5 μg of yellow fluorescent protein (YFP) plasmid using Lipofectamine 3000 (Life Technologies, Carlsbad, CA). Six hours after transfection, tsA201 HEK cells were plated on glass coverslips at 20% confluence and cultured for another 18 hours before patching.
Electrophysiology
Transfected tsA201 HEK cells were patched in the whole-cell configuration. WT and variant CaV3.2 currents were recorded in voltage-clamp mode using a Multiclamp 700B amplifier, a Digidata 1440A digital-to-analog converter, and pClamp10.4 software (Molecular Devices, Palo Alto, CA). Data were sampled at 10 kHz and filtered at 2 kHz. External modified Ringer’s solution contained (in mM): 105 NaCl, 1 MgCl2, 2 CaCl2, 5 KCl, 10 HEPES, and 10 Glucose, pH 7.4 with NaOH and 320 mOSM with D-sorbitol. Internal cesium gluconate pipette solution contained (in mM): 130 CsOH, 10 EGTA, 2 MgCl2, 7 NaCl, 10 HEPES, 3 ATP and 0.6 GTP, pH 7.25, and 300 mOsm with gluconic acid. Borosilicate glass pipettes were pulled with a PC-10 puller (Narashige) to a resistance of 4 mΩ. Access resistance was compensated to 80% and the capacitance of each cell was measured for current normalization.
Voltage data acquisition and analysis
A current-voltage (I-V) step protocol was used to determine voltage-dependent activation, current densities, activation rate, and inactivation rate properties for the CaV3.2WT and CaV3.2VAR channel populations, expressed in tsA201 HEK cells. The cell membrane potential was stepped from a resting voltage (Vr) of −90 mV to a test potential (Vt) of −70 mV for 150 ms then returned to the Vr for a 10-second interval to allow complete recovery from inactivation. The Vt was increased in 10-mV increments over a series of 11 steps; the last step was to + 30 mV. Peak inward Ca2+ currents (ICa) were plotted as a function of the Vt to generate I-V relations. The ICa was also normalized by the individual cell capacitance measurement for the comparison of current densities. Individual I-V and mean current-density-V relations were fit with the Boltzmann equation, where Ipeak = (Vt − Erev) G (1/1 + exp (Va,1/2 − Vt)/S)), and Erev is the reversal potential, Va,1/2 is the half-activation potential, G is the maximum slope conductance, and S is the slope factor that is inversely proportional to the effective gating charge. The ICa activation and inactivation phases at Vt = -30 mV were fit with single exponential functions to determine the tau of activation (τact) and the tau of inactivation (τinact).
A steady-state inactivation protocol was used to assess parameters of voltage-dependent inactivation; a Vr of −100 mV was stepped to a conditioning potential (Vc) between −100 and 0 mV for a duration of 1 second before a Vt of −30 mV was applied for 150 ms. Vc steps increased in 10-mV increments; between runs the cell membrane potential was returned to Vr for 10 seconds to allow for complete recovery from inactivation. ICa during Vt for each Vc was normalized to the peak ICa and fit with the Boltzmann relation: I/Imax = 1/(1 + exp (z*(Vc − Vi,1/2)/25.6)), where Vi,1/2 is the voltage of half-inactivation and z is the slope factor.
A tail-current protocol was used to determine deactivation rates; a short 2-ms prepulse to + 45 mV from a Vr of −100 mV was given to maximally open WT and mutant CaV3.2 channels without evoking inactivation before stepping the membrane potential of the cell back to a Vt of −100 mV. The deactivation phase of the large ICa evoked by Vt was fit with a single exponential function to determine the tau of deactivation (τdeact).
Statistical analyses
Data obtained with Clampex 10.4 were analyzed with Clampfit 10.4 (Molecular Devices, Sunnyvale, CA). Fits of the I-V and steady-state inactivation curves were done in SigmaPlot 12.5 (Jandel Scientific, San Rafael, CA). Student’s t-tests were employed to test significances of associations as indicated. Fisher’s exact test was used to test the association of carrier status and clinical phenotypes. The Mann-Whitney test was applied to compare COMPASS 31 scores between common haplotype carriers and noncarriers. Data are presented as the mean ± standard error. Statistical analysis was performed in SigmaPlot 12.5 (Jandel Scientific) and Prism (Graphpad, La Jolla, CA). Correction for multiple comparisons was not performed, given the small sample sizes.
Results
Three CACNA1H variants form a haplotype that frequently cosegregates with increased allelic TPSAB1 copies encoding α-tryptase
We first examined exome and genome sequences obtained from 12 families (87 individuals) with hereditary α-tryptasemia syndrome; all individuals in these families had increased allelic TPSAB1 copy number and multisystem complaints as described.1 All genes containing coding nonsynonymous segregating variants with a minor allele frequency (MAF) of at least 0.1 (in 1000 Genomes v3) in each family were identified, and compared to variant-containing genes that segregated in other families (GQ filter 20, allelic balance >25). Only six genes (CACNA1H, KCNJ12, MUC16, MUC6, PRAMEF1, and TPSG1), in addition to α-encoding variants mapping to TPSAB1 and TPSB2, were identified with segregating variants in five or more of the sequenced families (Supplementary Table S1 online). Of these, variants in TPSG1 and CACNA1H were identified in six families, with the same variants cosegregating in all individuals. The remaining variants were common in other databases or present in highly mutable genes (e.g., MUC16, MUC6) with little predicted effect on clinical phenotype.
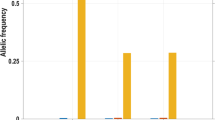
The MAF of the identified CACNA1H variants rs3751664 (NM_001005407:c.C2362T:p.(R788C)), rs58124832 (NM_001005407:c.G5594A:p.(R1865Q)), and rs72552056 (NM_001005407:c.C5995T:p.(R1999C)) were 0.089, 0.098, 0.091 in the ExAC database and 0.047, 0.031, 0.032 in the 1000 Genomes v3 database, respectively; the MAF for those identified in TPSG1, rs143120059 (NM_012467.3:c.G578A:p.(R193H)) and rs113856625 (NM_012467.3:c.C413T:p.(S138F)) were 0.053, 0.054 in the ExAC database and 0.025, 0.027 in the 1000 Genomes v3 database, respectively (Supplementary Table S2). Similar MAFs for variants within each gene, and our observation of coinheritance in all six families, suggested a larger shared haplotype was often co-inherited with TPSAB1 duplications or triplications (Figure 1a). Indeed, performing linkage disequilibrium analysis in 880 ClinSeq self-reported European-American individuals, we found that all five variants were present as a haplotype in 3.0% and were in linkage disequilibrium (Figure 1b, Supplementary Table S2); TPSAB1 duplications cannot be assessed using this method. Analysis of recombination rates at the tryptase locus confirmed two of the variants (rs58124832, rs72552056) in CACNA1H, and all tryptase encoding genes (TPSG1, TPSB2, TPSAB1, TSPD1) are within one large region with recombination rates of less than 3 cM/Mb, flanked by two relatively high recombination rate regions (left >25 cM/Mb and right >65 cM/Mb). These data suggest that this large region is more likely to be inherited as one haplotype in this population as well.

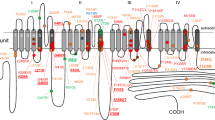
Location and protein changes in the CACNA1H variant haplotype near the tryptase locus. (a) Schematic of the105 kb region (GRCh38.p7) at 16p13.3, containing the tryptase locus (TPSG1, TPSAB1, TPSB2, and TPSD1) and CACNA1H. Haplotype variants identified in CACNA1H and TPSG1 are indicated; TPSAB1 is duplicated as seen in hereditary α-tryptasemia. (b) Recombination rate distribution for all variants in chromosome 16 (top) and of the region containing CACNA1H and TPSG1 (bottom) based upon 880 European-Americans in ClinSeq. CACNA1H, TPSG1, TPSB2, TPSAB1, TSPD1 regions are indicated in red, blue, pink, green, and purple, respectively. Haplotype variants (rs3751664, rs58124832, rs72552056) in CACNA1H are indicated by red vertical lines, and (rs143120059, rs113856625) in TPSG1 are blue vertical lines. (c) Schematic of CaV3.2 demonstrating locations of the amino acid substitutions (Δ) resulting from the three CACNA1H variants (red text in a).
Using real-time and digital droplet PCR (ddPCR), 46 families with hereditary α-tryptasemia syndrome (confirmed by clinical phenotype, elevated basal serum tryptase, and ddPCR demonstrating increased monoallelic α-tryptase encoding TPSAB1 copy number) were evaluated for the presence of the CACNA1H variant haplotype (CACNA1HVAR). A total of 32/46 families were found to have CACNA1HVAR cosegregating with increased TPSAB1 copy numbers. Eleven families did not carry any of the CACNA1H variants, and three families had two of the three variants (rs58124832 and rs72552056) segregating with increased TPSAB1 copy number. The TPSG1 variants were not detected in exome or genome sequence data in the absence of CACNA1H variants, and all 11 families without the haplotype were confirmed as carrying the major alleles at both TPSG1 loci. Only one individual carried the haplotype in trans with an α-tryptase duplication allele. Given the putative role of CaV3.2 in a number of clinical phenotypes of interest, we next focused our efforts on characterizing the CACNA1H variants and their effect on channel physiology.
The cosegregating CACNA1H haplotype variants impart partial gain of function in vitro
Expression of CACNA1HVAR results in smaller current densities than CACNA1HWT
The voltage-gated T-type calcium channel CaV3.2, encoded by CACNA1H, contains four transmembrane domains with interposed linker regions. The p.(R788C) amino acid substitution occurs in the domain I-II linker just proximal to the domain II transmembrane α-helix, and both p.(R1865Q) and p.(R1999C) substitutions are present in the carboxy terminus just distal to the domain IV transmembrane α-helix (Figure 1c). Both the domain I-II linker and the carboxy terminus contribute to voltage-dependent inactivation of T-type calcium channels;17, 18, 19, 20 thus we hypothesized that the loss of positively charged residues in these intracellular segments could alter fundamental channel properties.
Current densities, whole-cell ICa normalized for cell-membrane capacitance (pA/pF), differed between tsA201 HEK cells expressing wild-type CaV3.2 (CaV3.2WT) and variant (CaV3.2VAR) channels (P = 0.05; Figure 2a). At the peak of the I-V curve (−30 mV), current densities for CaV3.2WT were −27.5 pA/pF ± 5.1, n = 17, while CaV3.2VAR current densities were −17.1 pA/pF ± 3.3, n = 19. CaV3.2VAR may destabilize functional channel formation, resulting in the observed lower current densities. Differences in maximal conductance (G) measures obtained from I-V curve Boltzmann fits are also best explained as a consequence of reduced channel expression; CaV3.2WT G was 8.0 pS ± 1.1 (n = 17) while CaV3.2VAR G was 4.3 pS ± 0.6 (n = 19; P = 0.009) (Table 1).

Altered kinetics of CaV3.2VAR. (a) CaV3.2VAR has a smaller current density. Average current densities (pA/pF) as a function of voltage and fit using Goldman–Hodgkin–Katz (GHK) equation (values shown in Table 2). Current densities at −30 mV for CaV3.2WT (n = 17) and CaV3.2VAR (n = 19) were −27.5 ± 5.1 pA/pF, n = 17 and −17.1 ± 3.3 pA/pF, respectively; *P = 0.05. Data are shown as mean ± SEM, Student’s t-test. (b) CaV3.2VAR inactivates more slowly than CaV3.2WT. Voltages (top) used to evoke representative current (I) traces normalized to peak I at −30 mV (bottom); τinactivation obtained from single exponential fits of the inactivation phase was 20.1 ± 1.2 ms for CACNA1HWT (n = 17) compared to 25.8 ± 2.2 ms for CaV3.2VAR (n = 19); τactivation was 4.2 ± 0.4 ms for CACNA1HWT (n = 17) and 4.7 ± 0.7 ms for CaV3.2WT (n = 19). *P = 0.03, Student’s t-test. (c) CaV3.2VAR shifts voltage-dependent inactivation positively. Open probability-voltage relationships with Boltzmann fit indicated in solid lines; voltage of half-inactivation (Vi,1/2) were −60.6 ± 2.4 mV for CaV3.2WT (n = 13) and −55.3 ± 1.6 mV for CaV3.2VAR (n = 14), *P = 0.04; and slope factors (z) were 5.2 ± 0.4 for CaV3.2WT (n = 13) and 6.8 ± 0.4 for CaV3.2VAR (n = 14), P = 0.005. Data are shown as mean ± SEM, Student’s t-test. (d) CaV3.2VAR deactivates faster than CaV3.2WT. Voltages used (top) to evoke representative current (I) traces normalized to peak I at −100 mV (bottom); τdeactivation obtained from single exponential fits of the deactivation phase was 1.9 ± 0.2 ms for CaV3.2WT (n = 11) and 1.4 ± 0.08 ms for CaV3.2VAR (n = 12); *P = 0.02, Student’s t-test.
Properties of channel activation, namely the voltage sensitivity of activation (Va,1/2) and the rate of activation (τact), remained unaltered by the presence of CaV3.2VAR. CaV3.2WT Va,1/2 was −40.0 ± 2.0 mV, while CaV3.2VAR Va,1/2 was −38.9 ± 2 mV (P = 0.35). τact, determined through a single exponential fit of ICa development evoked by a voltage step from −90 to −30 mV, was 4.2 ± 0.4 ms for CaV3.2WT (n = 17) and 4.7 ± 0.7 ms for CaV3.2VAR (n = 19; P = 0.9).
CaV3.2VAR has altered inactivation properties compared to CaV3.2WT
τinact, the rate at which channels move into a non-conducting inactivated state, was determined through an exponential fit of the declining ICa evoked by membrane depolarization to −30 mV. CaV3.2VAR τinact was 25.8 ± 2.2 ms (n = 19), significantly slower than CaV3.2WT 20.1 ± 1.2 ms (n = 17; P = 0.03) (Figure 2b). A slower τinact would allow for greater calcium influx into neurons through conducting channels. CaV3.2VAR also altered parameters describing the voltage dependence of channel inactivation: the voltage of half-inactivation (Vi,1/2) and slope factor (z). Vi,1/2 was −60.6 ± 2.4 mV for CaV3.2WT (n = 13) and −55.3 ± 1.6 mV for CaV3.2VAR (n = 14; P = 0.04) (Figure 2c). Thus CaV3.2VAR would be predicted to allow greater calcium influx, as it would activate at the same low potentials as the CaV3.2WT population, but a greater depolarization would be required to place it in the nonconducting inactive state. z also differed significantly; it was 5.2 ± 0.4 for CaV3.2WT (n = 13) and 6.8 ± 0.4 for CaV3.2VAR (n = 14; P = 0.005). The steeper CaV3.2VAR z indicates that inactivation proceeds to completion over a smaller range of voltages than for CaV3.2WT channels.
CaV3.2VAR has altered deactivation properties compared to CaV3.2WT
τdeact, the rate at which channels move from the open to the closed state, differed significantly between CaV3.2WT and CaV3.2VAR channels (P = 0.02; Figure 2d). CaV3.2WT τdeact was 1.9 ± 0.2 ms (n = 11), while CaV3.2VAR τdeact was 1.4 ± 0.08 ms (n = 12); a faster τdeact attenuates calcium influx and promotes rapid neuronal repolarization.
Functional CACNA1H variant haplotype does not affect phenotypes among individuals with allelic TPSAB1 duplications
To determine potential clinical implications of the presence of the heterozygous CACNA1H partial gain-of-function variant allele, 57 individuals with TPSAB1 duplications were clinically phenotyped as described, with validated questionnaires for irritable bowel syndrome (Rome III criteria) and orthostatic symptoms (COMPASS 31, a validated measure of dysautonomic symptoms) being employed.1 Individuals with TPSAB1 triplications, those harboring two TPSAB1 duplication-carrying alleles, or those carrying the haplotype in trans with TPSAB1 duplications, were excluded from the analysis. Individuals with the CACNA1HVAR allele (n = 35), were compared to those found to carry the major allele for the three CACNA1H variants (CACNA1HWT; n = 22). Clinical phenotypic data were analyzed to assess potential impact upon the clinical phenotypes associated with TPSAB1 duplications. None of the features described in association with TPSAB1 duplications were significantly increased by the presence of CACNA1HVAR (Table 2; Supplementary Figure S1). CACNA1HVAR was associated with a modest but significant reduction in prevalence of systemic venom reactions (n = 2 v. 6; P = 0.04). Post-hoc analysis revealed that despite a small sample size, these data were powered to detect an approximately 30% difference for most parameters, thus if heterozygous expression of the CACNA1HVAR allele had an effect on clinical phenotype, it did not appear to be a major contributor.
In the absence of hereditary α-tryptasemia, the CACNA1H variant haplotype does not alter the prevalence of symptoms associated with TPSAB1 duplications
It remained possible that any modifying effect of CACNA1HVAR on assessed clinical phenotypes was not detected because of the dominant effect of increased TPSAB1 copy number, as suggested by the observed gene-dose effect in hereditary α-tryptasemia syndrome.1 Therefore, we next looked for an effect of the CACNA1HVAR haplotype (defined by the presence of the three CACNA1H variants) on the same set of clinical symptoms, within the ClinSeq cohort, an unselected population of healthy unrelated adults.10 After stringent filtering was applied, requiring at least 10 × coverage over all three variants, 22 individuals were identified as having the CACNA1HVAR allele. Some individuals with the variant haplotype may have been classified as carrying only one or two variants, owing to inadequate exon capture in some samples; however, based upon the calculated frequency of the haplotype, we expected to identify only 26 individuals with the haplotype (Supplementary Table S2). Using the same selection strategy, an additional 50 individuals with wild-type CACNA1H sequence at these loci were also selected at random, and attempts were made to contact all individuals for a blinded phone interview.
Basal serum tryptase levels were obtained on all individuals to screen for hereditary α-tryptasemia, and ddPCR was performed to confirm genetic diagnosis in all individuals with tryptase >8 ng/ml. Of the 22 individuals with CACNA1HVAR, six were subsequently excluded, owing to the identification of increased monoallelic TPSAB1 copy, and three were not available for phone interview. Four of the randomly selected CACNA1HWT individuals were likewise unavailable for interview. The remaining 13 individuals with the CACNA1HVAR haplotype were then compared to the 46 randomly selected individuals who both had wild-type CACNA1H sequence (CACNA1HWT) and did not harbor TPSAB1 duplications. The age and demographic characteristics did not differ significantly between these two populations. No association was seen between the variant haplotype and any of the clinical features associated with TPSAB1 duplications: including irritable bowel syndrome (by Rome III criteria), orthostatic symptoms (by COMPASS 31) or systemic reactions to venom (Table 3, Supplementary Figure S1).
Discussion
A previously unrecognized haplotype containing three functional variants in LD within CACNA1H is frequently coinherited on the same allele as additional α-tryptase-encoding TPSAB1 copies. This haplotype appears to be common in Caucasians and results in a partial gain of function in vitro.
Many clinical symptoms associated with hereditary α-tryptasemia are variably expressed. We had speculated that in the syndromic presentation, a second coinherited genetic variant might contribute to the observed differences in expressivity, as may occur relatively commonly in single gene disorders.17 Given the frequent coinheritance of this functional CACNA1HVAR haplotype and the physiology attributed to CaV3.2 signaling, a compelling case can be made for a contribution to the multisystem complaints seen in this syndrome. Despite this, in the heterozygous state, we were unable to detect any clinical effects of the CACNA1HVAR haplotype on manifestations of hereditary α-tryptasemia syndrome among individuals with TPSAB1 duplications or from the ClinSeq cohort. However, it remains possible that individuals homozygous for this functional haplotype may display a clinical phenotype.
The potential clinical importance of the newly discovered CACNA1HVAR haplotype beyond symptoms associated with increased TPSAB1 copy number remains unexplored. The p.(R788C) and p.(R1865Q) amino acid substitutions have been linked to neurological disorders—childhood absence epilepsy6,18 and autism spectrum disorders,19 respectively—and a distinct gain-of-function mutation resulting from the missense c.4645A>G:p.(M1549V) substitution has been associated with primary hyperaldosteronism.5 While these phenotypes were not reported in the small sample we studied, it remains possible that this CACNA1HVAR haplotype may have significant impacts on these or other clinical phenotypes.
It also is important to note that in the model system employed to study CaV3.2 channel physiology, only wild-type or variant proteins were overexpressed. Thus, homozygosity of the variant allele might be required in vivo to impart a clinical phenotype; we have confirmed only seven such individuals, all in association with increased TPSAB1 copy number. Of these, four individuals each had two alleles with duplications, and three each had one duplicated and one triplicated allele, making any comparison between groups anecdotal, and any rational statistical analysis impractical. All individuals homozygous for increased TPSAB1 copy number (n = 7) identified to date have been symptomatic and also homozygous for the CACNA1HVAR haplotype.
Two coding nonsynonymous TPSG1 variants were also present exclusively with the CACNA1HVAR haplotype in all 46 families. These variants are frequently encountered as part of this haplotype as well, and may also have a functional effect on mast-cell physiology. γ-Tryptase, encoded by TPSG1, has a similar structure and is believed to have a similar proteolytic function to α- and β-tryptases, albeit a more limited one. It is not known to be secreted, rather transiently expressed on the surfaces of mast cells following degranulation.20 While potential effects of these variants have not yet been investigated in vitro—a considerable challenge in the human system, given the limited data on the physiology of γ-tryptase and the confounding presence of secreted α- and β-tryptases in these same systems—any major contribution to common clinical phenotypes associated with hereditary α-tryptasemia syndrome, if present, would also have been detected in this analysis. Much additional work is required to understand the functional role of wild-type γ-tryptase in human mast cells before functional analysis of these variants can be undertaken.
The remaining four genes excluded from functional analysis were not found to have any variant shared by more than three families within the cohort (Supplementary Table S1). Further, tissue expression of these genes is highly restricted: KCNJ12 has been reported only in skeletal muscle,21 MUC6 and MUC16 encode mucins restricted to the gastric mucosa22 and ovarian/ocular tissues respectively,23,24 and PRAMEF1 is a cancer/testis antigen.25 Of these, only KCNJ12 variants might be predicted to result in a phenotype associated with hereditary α-tryptasemia syndrome. KCNJ12 encodes an inward rectifying potassium channel, mutations in which can lead to periodic paralysis.21 However, the rarest KCNJ12 variant in our cohort was common (ExAC MAF 0.18), making any variant unlikely to be damaging or result in observed clinical phenotypes.
As additional patients with hereditary α-tryptasemia are identified, expanded interrogation of phenotypes among individuals homozygous for the CACNA1HVAR haplotype may yet unmask clinical effects of associated alterations in CaV3.2 signaling.
References
Lyons JJ, Yu X, Hughes JD et al. Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nat Genet 2016;48:1564–1569.
Itan Y, Shang L, Boisson B et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc Natl Acad Sci USA 2015;112:13615–13620.
Marger F, Gelot A, Alloui A et al. T-type calcium channels contribute to colonic hypersensitivity in a rat model of irritable bowel syndrome. Proc Natl Acad Sci USA 2011;108:11268–11273.
Gangarossa G, Laffray S, Bourinet E, Valjent E . T-type calcium channel Cav3.2 deficient mice show elevated anxiety, impaired memory and reduced sensitivity to psychostimulants. Front Behav Neurosci 2014;8:92.
Scholl UI, Stolting G, Nelson-Williams C et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife 2015;4:e06315.
Khosravani H, Altier C, Simms B et al. Gating effects of mutations in the Cav3.2 T-type calcium channel associated with childhood absence epilepsy. J Biol Chem 2004;279:9681–9684.
Wang XL, Tian B, Huang Y et al. Hydrogen sulfide–induced itch requires activation of Cav3.2 T-type calcium channel in mice. Sci Rep 2015;5:16768.
Garcia-Caballero A, Gadotti VM, Stemkowski P et al. The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron 2014;83:1144–1158.
Choi S, Na HS, Kim J et al. Attenuated pain responses in mice lacking Ca(V)3.2 T-type channels. Genes Brain Behav 2007;6:425–431.
Biesecker LG, Mullikin JC, Facio FM et al. The ClinSeq Project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res 2009;19:1665–1674.
Zhang Y, Yu X, Ichikawa M et al. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol 2014;133:1400–1409.e1-5.
Brennan CW, Verhaak RG, McKenna A et al. The somatic genomic landscape of glioblastoma. Cell 2013;155:462–477.
Johnston JJ, Lewis KL, Ng D et al. Individualized iterative phenotyping for genome-wide analysis of loss-of-function mutations. Am J Hum Genet 2015;96:913–925.
Barrett JC, Fry B, Maller J, Daly MJ . Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005;21:263–265.
Sletten DM, Suarez GA, Low PA, Mandrekar J, Singer W . COMPASS 31: a refined and abbreviated Composite Autonomic Symptom Score. Mayo Clin Proc 2012;87:1196–1201.
Drossman DA, Dumitrascu DL . Rome III: New standard for functional gastrointestinal disorders. J Gastrointestin Liver Dis 2006;15:237–241.
Soldatov NM, Zuhlke RD, Bouron A, Reuter H . Molecular structures involved in L-type calcium channel inactivation. Role of the carboxyl-terminal region encoded by exons 40–42 in alpha1C subunit in the kinetics and Ca2+ dependence of inactivation. J Biol Chem 1997;272:3560–3566.
Stotz SC, Hamid J, Spaetgens RL, Jarvis SE, Zamponi GW . Fast inactivation of voltage-dependent calcium channels. A hinged-lid mechanism? J Biol Chem 2000;275:24575–24582.
Kobrinsky E, Schwartz E, Abernethy DR, Soldatov NM . Voltage-gated mobility of the Ca2+ channel cytoplasmic tails and its regulatory role. J Biol Chem 2003;278:5021–5028.
Vitko I, Bidaud I, Arias JM, Mezghrani A, Lory P, Perez-Reyes E . The I-II loop controls plasma membrane expression and gating of Ca(v)3.2 T-type Ca2+ channels: a paradigm for childhood absence epilepsy mutations. J Neurosci 2007;27:322–330.
Posey JE, Harel T, Liu P et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med 2017;376:21–31.
Vitko I, Chen Y, Arias JM, Shen Y, Wu XR, Perez-Reyes E . Functional characterization and neuronal modeling of the effects of childhood absence epilepsy variants of CACNA1H, a T-type calcium channel. J Neurosci 2005;25:4844–4855.
Splawski I, Yoo DS, Stotz SC, Cherry A, Clapham DE, Keating MT . CACNA1H mutations in autism spectrum disorders. J Biol Chem 2006;281:22085–22091.
Caughey GH . Tryptase genetics and anaphylaxis. J Allergy Clin Immunol 2006;117:1411–1414.
Ryan DP, da Silva MR, Soong TW et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell 2010;140:88–98.
De Bolos C, Garrido M, Real FX . MUC6 apomucin shows a distinct normal tissue distribution that correlates with Lewis antigen expression in the human stomach. Gastroenterology 1995;109:723–734.
Yin BW, Lloyd KO . Molecular cloning of the CA125 ovarian cancer antigen: identification as a new mucin, MUC16. J Biol Chem 2001;276:27371–27375.
Argueso P, Spurr-Michaud S, Russo CL, Tisdale A, Gipson IK . MUC16 mucin is expressed by the human ocular surface epithelia and carries the H185 carbohydrate epitope. Invest Ophthalmol Vis Sci 2003;44:2487–2495.
Hermes N, Kewitz S, Staege MS . Preferentially expressed antigen in melanoma (PRAME) and the PRAME family of leucine-rich repeat proteins. Curr Cancer Drug Targets 2016;16:400–414.
Acknowledgements
This study was supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases and of the National Human Genome Research Institute, National Institutes of Health, the NIAID Clinical Genomics Program, and funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. Funding to J.J.L. was also provided in part by ARTrust/The Mastocytosis Society Research Award in Mastocytosis and/or Mast Cell Activation Syndrome. Funding to M.A.C.was provided by the Canadian Institutes of Health Research. L.G.B. and K.L.L. were supported by the Intramural Research Program of the National Human Genome Research Institute, grants HG200359 08 and HG200387 03. The investigators thank the patients, their families, and healthy volunteers who contributed to this research, as well as the clinical staff of the LAD and NHGRI for their contributions.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Author contributions
J.J.L., S.C.S., M.A.C., and J.D.M. designed the study. J.J.L., J.D.M, C.N., T.D., N.J., all recruited subjects to the study. J.J.L., J.D.M., and J.C., collected and/or analyzed clinical data. X.Y., J.D.H., C.H., Y.Z. A.J.O., J.J.M., L.G.B., and J.D.M. performed and supported genomic sequencing and X.Y., J.D.H., C.H., Y.Z., and A.J.O. performed the bioinformatics analyses. J.C. and Y.L. performed, and J.J.L. designed and performed PCR assays. S.C.S. and M.A.C. designed and performed the functional electrophysiology studies. K.L.L. and L.G.B. facilitated all ClinSeq related study data and H.S. performed LD calculations and analysis. S.C.S., J.J.L., M.A.C, and J.D.M. prepared the draft manuscript. All authors contributed to discussion of the results and to manuscript preparation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
L.G.B. is an uncompensated adviser to Illumina, receives royalties from Genentech and Amgen, and receives honoraria from Wiley-Blackwell. The other authors declare no conflict of interest.
Additional information
The first two authors contributed equally to this work.
Supplementary material is linked to the online version of the paper at
Supplementary information
Rights and permissions
About this article

Cite this article
Lyons, J., Stotz, S., Chovanec, J. et al. A common haplotype containing functional CACNA1H variants is frequently coinherited with increased TPSAB1 copy number. Genet Med 20, 503–512 (2018). https://doi.org/10.1038/gim.2017.136
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.136
Keywords
This article is cited by
-
Reversible Elevation of Tryptase Over the Individual's Baseline: Why is It the Best Biomarker for Severe Systemic Mast Cell Activation and MCAS?
Current Allergy and Asthma Reports (2024)
-
Hereditary Alpha-Tryptasemia: a Commonly Inherited Modifier of Anaphylaxis
Current Allergy and Asthma Reports (2021)
