
Abstract
Purpose:
To investigate pan-ethnic SMN1 copy-number and sequence variation by hybridization-based target enrichment coupled with massively parallel sequencing or next-generation sequencing (NGS).
Methods:
NGS reads aligned to SMN1 and SMN2 exon 7 were quantified to determine the total combined copy number of SMN1 and SMN2. The ratio of SMN1 to SMN2 was calculated based on a single-nucleotide difference that distinguishes the two genes. SMN1 copy-number results were compared between the NGS and quantitative polymerase chain reaction and/or multiplex ligation-dependent probe amplification. The NGS data set was also queried for the g.27134T>G single-nucleotide polymorphism (SNP) and other SMN1 sequence pathogenic variants.
Results:
The sensitivity of the test to detect spinal muscular atrophy (SMA) carriers with one copy of SMN1 was 100% (95% confidence interval (CI): 95.9–100%; n = 90) and specificity was 99.6% (95% CI: 99.4–99.7%; n = 6,648). Detection of the g.27134T>G SNP by NGS was 100% concordant with an restriction fragment-length polymorphism method (n = 493). Ten single-nucleotide variants in SMN1 were detectable by NGS and confirmed by gene-specific amplicon-based sequencing. This comprehensive approach yielded SMA carrier detection rates of 90.3–95.0% in five ethnic groups studied.
Conclusion:
We have developed a novel, comprehensive SMN1 copy-number and sequence variant analysis method by NGS that demonstrated improved SMA carrier detection rates across the entire population examined.
Genet Med advance online publication 19 January 2017
Similar content being viewed by others


Introduction
Spinal muscular atrophy (SMA; MIM 253300) is a neuromuscular disorder caused by loss of motor neurons in the spinal cord and brainstem, leading to generalized muscle weakness and atrophy that impairs activities such as crawling, walking, sitting up, and controlling head movement.1 SMA has variable expressivity, with a broad range of onset and severity. In severe cases, death occurs within the first 2 years of life, due mostly to respiratory failure.2 It has an incidence of about 1 in 10,000 live births and a carrier frequency of about 1/40 to 1/100 in different ethnic groups, with a higher carrier frequency among Caucasians and lower carrier frequencies among African Americans and Hispanics.3,4,5,6 SMA is caused by biallelic mutations in the survival motor neuron 1 (SMN1) gene, including deletions, gene conversions, and intragenic mutations, whereas SMN2 copy number may modify disease severity.7 SMN1 and SMN2 are highly homologous differing in five base pairs, none of which changes the amino acid sequence. A single C-to-T change in SMN2 exon 7 (c.840C>T) affects an exonic splicing enhancer, which results in a reduction of full-length transcripts from SMN2.8 This nucleotide is considered the only functional paralogous sequence variant9 (PSV; Figure 1a ) and is what differentiates SMN1 from SMN2.

The SMN1 and SMN2 next-generation sequencing sequence alignment surrounding the functional paralogous sequence variant (PSV) at c.840. (a) The SMN gene PSV1 (c.840C/T), PSV2 (c.888 + 100A/G) and SMN1 single-nucleotide polymorphism (SNP) g.27134T>G are located within a 148-bp region spanning exon 7 and intron 7 of the SMN1 or SMN2 gene. (b) The alignment of pair-end sequence reads (2 × 100) in a normal and SMN1/SMN2 gene hybrid sample. The red or purple box represents the pair-end read R1 or R2, respectively. The green letters at the PSV1, PSV2, or the SMN1 SNP loci indicate that the aligned reads match the reference sequence at these positions. Yellow letters indicate the mismatched bases in the correctly aligned reads caused by sequence polymorphism or a gene conversion event. Red letters indicate the mismatched bases in the misaligned reads caused by sequence polymorphism or gene conversion. (c) Sequence pileups of read pairs at the correct SMN1 locus (top) and incorrect SMN2 locus (bottom).
SMA has features that can be recognized clinically, but molecular testing is typically required to confirm the diagnosis. Polymerase chain reaction (PCR) coupled with restriction fragment-length polymorphism analysis is a commonly used diagnostic test for SMA,10 but this method does not detect carrier status. The first carrier test for SMA, developed in 1997, used a competitive PCR strategy for quantification of SMN1 copy number.11 Since then, the development of higher-throughput methods, such as multiplex ligation-dependent probe amplification (MLPA) and quantitative PCR (qPCR), has enabled SMA carrier screening on a population basis.12,13 These methodologies determine SMN1 copy number by interrogating the c.840C/T functional PSV that distinguishes the two SMN genes.
Massively parallel sequencing or next-generation sequencing (NGS) technologies have rapidly transformed medicine as cost-effective approaches to detecting pathogenic variants on a genomic scale in patients with genetic diseases.14 Recently developed NGS-based carrier screening panels offer increased detection rates relative to conventional genotyping in a high-throughput mode for a large number of genes.15,16 In addition, NGS is now used on a clinical basis for the detection of copy-number variants (CNVs).17,18 The ability to detect such pathogenic variants when performing carrier screening using NGS is particularly important for diseases in which a high percentage of pathogenic variants are CNVs, as is the case with SMA. However, NGS-based CNV detection is challenging for deletions and duplications at the single exon or subexon level because of technical noise introduced by uneven coverage in regions with variable GC content, nonlinear amplification by PCR, and/or inter-run variations caused by assay artifacts known as batch effects. Another major drawback of CNV analysis by short-read NGS is the lack of locus-specific computational programs for genes with highly homologous sequences that are not easily mapped to the genome. These genes, including SMN1 and SMN2, are normally excluded from NGS variant-calling and copy-number analyses.19 In addition, SMN1 and SMN2 often undergo gene conversion events leading to gene hybrids that harbor PSVs from both genes.20 This complicates CNV analysis by NGS and underscores the need for nuanced data analysis to avoid errors caused by misalignment and gene conversion. SMN1 copy-number analysis using a Bayesian hierarchical model applied to the 1000 Genomes Project database was recently reported.21 This analysis characterized individuals as “likely,” “possibly,” or “unlikely” SMA carriers. However, to our knowledge, an NGS-based clinical method for copy-number analysis of SMN1 and/or other genes with highly homologous sequences has not been reported in the literature.
Sequence variants including single-nucleotide variants or other small deletions, insertions, or insertions/deletions in SMN1 are medically relevant but not routinely detected by existing SMA carrier testing approaches. A recent study identified a SNP (g.27134T>G) tightly linked to a haplotype in silent carriers who have two copies of SMN1 on one chromosome and zero copies on the other chromosome (2 + 0 configuration) in certain populations.22 Analysis of this SNP was recommended in a recent update on SMA carrier testing by the American College of Medical Genetics and Genomics.23 In addition, while whole-gene or exonic CNVs account for the majority of SMA disease alleles, approximately 2.5% of SMA pathogenic variants are point mutations.6 These pathogenic single-nucleotide variants are not detected by carrier testing methods that only interrogate the c.840 PSV.
We developed a novel method called paralogous gene copy-number analysis by ratio and sum (PGCNARS) for SMA carrier testing based on short-read NGS data. This method was rigorously validated in a clinical setting using 6,738 pan-ethnic samples and compared to results generated by MLPA or qPCR. In addition, the g.27134T>G SNP associated with 2 + 0 SMA carrier status and pathogenic SMN1 sequence variants were also analyzed.
Materials and Methods
DNA samples
The analyses were performed using de-identified samples submitted to Baylor Genetics Laboratory for carrier testing for a panel of diseases, including SMA, by NGS, qPCR, and MLPA with the approval from the institutional review board at Baylor College of Medicine. DNA was extracted from whole blood using commercially available DNA isolation kits (Gentra Systems, Minneapolis, MN), following the manufacturer’s instructions.
SMN1 copy-number analysis by MLPA
SMN1 copy number was analyzed using the MCR-Holland SALSA MLPA Kit P060-B2 (MRC-Holland, Amsterdam, The Netherlands) or custom-designed MLPA reagents, according to the manufacturer’s recommendations. The MLPA reagent contains sequence-specific probes targeted to exons 7 and 8 of both SMN1 and SMN2 (ref. 24). The MLPA data were analyzed using Coffalyzer software (MRC-Holland).
SMN1 copy analysis by TaqMan qPCR
SMN1 copy number was assessed using the TaqMan qPCR assay as part of a panel using the BioMark 96.96 Dynamic Array (Fluidigm, South San Francisco, CA). Exon 7 from both the SMN1 and SMN2 genes was amplified by the following primer pair: 5′-ATAGCTATTTTTTTTAACTTCCTTTATTTTCC-3′ and 5′-TGAGCACCTTCCTTCTTTTTGA-3′. A probe that specifically targets the SMN1 PSV (FAM-TTGTCTGAAACCCTG) was used to detect SMN1, whereas SMN2 was blocked by a probe that targets the SMN2 PSV (VIC-TTTTGTCTAAAACCC). qPCR was performed on the BioMark HD system (Fluidigm) as previously described, with minor modifications.25 Copy number was calculated using the ▵▵Ct method by normalizing to the genomic reference of the case and to the batch reference within the chip.26
Capture enrichment and NGS
A previously described protocol27 using capture-based target enrichment followed by NGS was adapted for the clinical test of 158 genes, including SMN1, selected for carrier testing. Briefly, genomic DNA was fragmented by sonication, ligated to multiplexing paired-end adapters (Illumina, San Diego, CA), amplified by PCR with indexed (barcoded) primers for sequencing, and hybridized to biotin-labeled, custom-designed capture probes (NimbleGen; Roche, Madison, WI) in a solution-based reaction. Hybridization was performed at 47 °C for at least 16 h, followed by paired-end sequencing (100 bp) on the Illumina HiSeq 2500 platform, with average coverage of >300× in the targeted regions.
NGS data processing and data quality control
Raw-image data conversion and demultiplexing were performed following Illumina’s primary data analysis pipeline using CASAVA version 2.0 (Illumina). Low-quality reads (Phred score <Q25) were removed before demultiplexing. Batched samples from the same capture pool were grouped and processed together. Sequences were aligned to the hg19 reference genome by NextGENe software (SoftGenetics, State College, PA) using the recommended standard settings for single-nucleotide variant and insertion/deletion discovery. In every sample, the average coverage depth of each targeted exon of nonhomologous genes was extracted and normalized according to our previously published methods.18 Similar to derivative log ratio spread used in the quality assurance of array-based comparative genomic hybridization data analysis, derivative ratio spread, defined below, was used to quantify the coverage depth variation of each sample from the NGS data.

where δ represents the difference of normalized coverage ratio between two adjacent exons; µ is the mean of all δ; N is the total number of data points (the number of total exons minus 1). A sample with derivative ratio spread > 0.1 is considered to have not passed quality control and therefore was not included in the copy-number analysis. The script for the detection of SMN1 copy numbers using next-generation sequencing coverage depth is deposited at https://sourceforge.net/projects/PGCNARS.
Results
SMN1 and SMN2 NGS sequence alignment based on the functional PSV at c.840
Since SMN1 and SMN2 differ at only five bases, most of the SMN1- or SMN2-derived NGS reads (2 × 100-bp pair-end (PE) sequencing used in this work) were indistinguishable. As a result, these reads were ambiguously aligned to either SMN1 or SMN2 with poor mapping quality, making read depth–based copy-number analysis inapplicable. Notably, reads containing at least one SMN1 or SMN2 PSV were mapped to the reference locus with higher mapping specificity. For example, in a sample with two copies of SMN1 and zero copies of SMN2 determined by MLPA, all correctly mapped NGS reads contained the SMN1 PSV (c.840C) in exon 7 (Supplementary Figure S1a online). Reads that mapped incorrectly to exon 7 of SMN2 were those without the SMN1 PSV (Supplementary Figure S1b online).
Effects of SMN1 and SMN2 gene conversion on sequence alignment and read-depth analysis
Since the functional PSV at c.840 is the only base that can be reliably used to differentiate the SMN1 and SMN2 genes, accurate read-depth data at this locus are necessary to determine the SMN1 and SMN2 copy number. However, gene conversions can produce SMN1 and SMN2 gene hybrids that harbor both SMN1 and SMN2 PSVs in a single SMN gene. In these samples, the SMN1 gene-specific functional PSV (PSV1; c.840C in SMN1) and the SMN2 PSV (PSV2; c.888 + 100G in SMN2) can be found in a haplotype block containing exon 7 and intron 7 ( Figure 1a ). The NGS reads derived from such gene hybrid regions may confound the mapping algorithm and result in incorrect alignment ( Figure 1b ). For example, in a gene hybrid sample with the SMN1 functional PSV (c.840C), SMN1 SNP (g.271347T>G), and SMN2 PSV (c.888 + 100G) present in cis, 26% of the SMN1 sequences with the functional PSV mapped to the SMN2 locus ( Figure 1c ). These SMN1 reads were misaligned to SMN2 because the PE read-mapping algorithm did not always utilize the functional PSV c.840C to anchor the read pairs to the SMN1 locus when the SMN1 PSV c.840C was present on the first read (R1) and the SMN2 intronic PSV and the SMN1 SNP were present on the second read (R2). Therefore, we decoupled the 2 × 100 PE reads and performed alignment based on single-end reads to achieve more accurate read-depth data at the SMN functional PSV locus. This was an essential step to correctly map reads containing the c.840C PSV to the SMN1 gene. We compared the performance of PE and single-end alignment for eight gene-hybrid samples with three copies of SMN1 and one copy of SMN2 confirmed by MLPA. We found that single-end mapping was more accurate for SMN gene copy-number analysis. Compared with the single-end alignment method, SMN1 to SMN2 copy-number ratio was decreased and SMN1 copy number was underestimated by the PE alignment because some of the SMN1 reads were misaligned to the SMN2 locus (Supplementary Figure S2 online).
Calculation of SMN1 and SMN2 copy number by the ratio and sum of their NGS reads
To determine SMN1 and SMN2 copy number using NGS data, we first hypothesized that in any given sample, the SMN1 to SMN2 copy-number ratio should be determined by their gene-specific reads ratio. To test this hypothesis, we calculated the SMN to SMN2 copy-number ratio for all samples in this study (n = 6,738) by surveying informative reads harboring the c.840C/T functional PSV in exon 7 or the c.*233T/A PSV in exon 8. The samples fell into three major populations with SMN1 to SMN2 copy-number ratios of one, two, or three (Supplementary Figure S3 online). This observation was in line with the fact that the most common configurations of SMN1 and SMN2 include individuals with two copies of SMN1 and two copies of SMN2, two copies of SMN1 and one copy of SMN2, or three copies of SMN1 and one copy of SMN2 (refs. 28–30). Samples with zero copies of SMN2 were also relatively common (Supplementary Figure S3 online). Samples with the same SMN1 and SMN2 gene copy-number ratio frequently had different absolute gene copy numbers (e.g., individuals with two copies of SMN1 and SMN2 and those with three copies of each). Therefore, the copy-number ratio itself could not be used directly to infer SMN1 and SMN2 copy number; it was informative only when it was used together with the combined SMN1 and SMN2 total copy number.
We then calculated SMN1 and SMN2 total copy number using read-depth data using our previously published NGS-based copy-number analysis method, with modifications.18 We made an important adjustment to the published protocol, which was to perform the analysis by capture batch. Samples pooled together in a single hybridization-based target enrichment reaction were analyzed and normalized as a group. This approach reduced the batch effects introduced by target capture, PCR after capture, and sequencing variation. We observed a significantly higher error rate for SMN1 copy-number calculations when samples from different capture pools were analyzed together, even when they were sequenced in the same flow-cell (Supplementary Table S1 online).
To calculate SMN1 and SMN2 total copy number, we normalized exonic read-depth to total mapped reads of all targeted genes included in our carrier screening panel. All reads aligned to either SMN1 or SMN2 were counted in this step, including both gene-specific reads and those nondistinguishing reads lacking PSVs. Next, samples with SMN1 to SMN2 copy-number ratios between 0.8 and 1.2 were grouped together to identify the median sample, which generally was a sample with two copies each of SMN1 and SMN2. The median sample served as an intrabatch SMN1 and SMN2 total read-depth normalizer for subsequent calculations. The exact SMN1 and SMN2 copy number of this normalizer was confirmed by MLPA or qPCR and demonstrated complete concordance with the NGS-predicted value (i.e., two copies of SMN1 and SMN2) in >50 consecutive batches. Finally, the SMN1 copy number for each sample was determined by applying the formula

in which n1 is the calculated copy number of SMN1; rd1 and rd2 are the read depths of the c.840 PSV at SMN1 and SMN2, respectively; Σc is the combined exonic (exon 7) coverage of SMN1 and SMN2; and χc is the median of all the calculated Σc in a group of samples batched together for the analysis. The overall SMN1 and SMN2 copy-number calculation algorithm is illustrated in Figure 2 . Note that the formula can also be used to compare the exon 8 copy-number analysis with the exon 7 copy-number results by applying the coverage data of the exon 8 PSV (c.*233T/A). Using this method, we were able to differentiate SMA carriers who had one copy of SMN1 and SMN2 (1/1) from noncarriers who had two copies of each (2/2), although their SMN1 to SMN2 copy-number ratios were not distinguishable (Supplementary Table S2 online). Individuals with 1/1 and 2/2 had an average of 2.1 and 3.98 total SMN1 and SMN2 copy numbers, respectively. The same principle was applied to distinguish 1/2 carriers from 2/3 carriers and/or other similar configurations.

A novel computational algorithm PGCNARS (paralogous gene copy-number analysis by ratio and sum) for SMN1 copy-number analysis using next-generation sequencing coverage depth data for spinal muscular atrophy carrier screening. PGCNARS involves three major steps for the SMN1 copy-number analysis. First, for each sample in the same capture pool, the copy-number ratio of SMN1 to SMN2 is calculated using the read depth of the paralogous sequence variants in exon 7 (c.840C/T) or exon 8 (c.*233T/A) of SMN1 and SMN2 (step a1–3). The SMN1 and SMN2 total copy number was determined by their exonic coverage data after normalization to the read depth of the median identified in the sample group (step b1–7). Finally, the SMN1 copy number in each sample is calculated based on the SMN1 to SMN2 copy-number ratio and their total copy number (step c).
Reproducibility, sensitivity, and specificity of SMN1 copy-number analysis
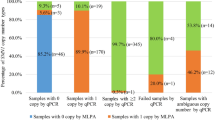
To determine the reproducibility of this new NGS-based copy-number analysis for SMN1, 68 samples were repeated in three independent runs; among these, 53 samples had two copies of SMN1, 11 had three or more copies of SMN1, and 4 had one copy of SMN1. This reproducibility test demonstrated complete concordance for all samples in all three runs. Next we analyzed 6,738 clinical samples submitted to our laboratory for carrier testing by comparing the qPCR and/or MLPA results with those generated by PGCNARS ( Table 1 ). The test sensitivity was 100% for SMA carriers (95% CI: 95.9–100%; n = 90) with a test specificity at 99.6% (95% CI: 99.4–99.7%; n = 6,648). For samples with two copies of SMN1, the NGS method’s test sensitivity and specificity were 99.4% (95% CI: 99.1–99.5%; n = 5,480) and 98.3% (95% CI: 97.5–98.9%; n = 1,258), respectively. For samples with three or more copies of SMN1, test sensitivity and specificity were 98.2% (95% CI: 97.3–98.8%; n = 1,168) and 99.8% (95% CI: 99.7–99.9%; n = 5,570), respectively. To test whether the NGS-based SMN1 copy-number analysis can be used for the diagnosis of patients with SMA, we tested a familial tetrad in which two children were affected by SMA. Our NGS analyses showed that both of the affected children had zero copies of SMN1, while their parents were carriers with one copy of SMN1 (Supplementary Figure S4 online).
Multiethnic SMN1 copy-number analysis for SMA carrier population screening by NGS
The multiethnic SMN1 copy-number analysis data for SMA carrier population screening by NGS is summarized in Table 2 . In 5,344 individuals with known ethnicity, African Americans and Hispanics had the lowest carrier frequencies with SMN1 deletion, at 1.0 and 0.9%, whereas Asians had the highest carrier frequency at 2.4%. Caucasians and individuals of Ashkenazi Jewish ancestry had SMA carrier frequencies at 1.4 and 1.9%, respectively. About 47.8% of African Americans had three or more copies of SMN1, which is significantly higher than any other population. These results are consistent with previous studies of SMN1 copy-number distribution in the general population,4 indicating that the NGS method reported herein is robust in its determination of SMN1 copy number.
Detection of the g.27134T>G SNP associated with 2 + 0 SMA carrier status by NGS
Next we tested whether our NGS assay could detect a recently identified g.27134T>G SNP associated with 2 + 0 SMA carrier status.22 Our NGS method to call the g.27134T>G SNP yielded results completely concordant with those generated by a restriction fragment-length polymorphism assay in 493 consecutive samples (Supporting Information and Supplementary Methods and Procedures online; Supplementary Figures S6 and S7 online; Supplementary Tables S4 and S5 online). Importantly, using the NGS method, we found that 574 of the 956 individuals (79%) with three or more copies of SMN1 were also positive for the g.27134T>G SNP, whereas only 5% of individuals with two copies of SMN1 were carriers of the g.27134T>G SNP ( Table 2 ). Therefore, testing for this SNP in the general population could theoretically identify 2 + 0 SMA carriers. In our cohort, linkage of the SNP with the SMN1 duplicated allele varied by ethnic group. Based on the configurations of SMN1 copy number and the g.27134T>G SNP genotype, we found that linkage was the highest among African Americans; 74.5% of duplicated SMN1 alleles were also positive for the g.27134T>G SNP. Linkage was the lowest for Asians, with a positive SNP frequency of 6.7% among the duplicated alleles. The linkage was 12.3, 35.3, and 33.3% for Caucasians, Hispanics, and Ashkenazi Jews, respectively. When SMN1 copy-number and g.27134T>G SNP analyses were combined to identify SMA carriers, the detection rate was increased to 90.3–95.0% in different ethnic groups compared with SMN1 copy number–based carrier testing ( Table 3 ). Therefore, the residual risk of being an SMA carrier after a negative screening result (i.e., two copies of SMN1 and negative for g.27134T>G SNP) decreases in all populations ( Table 3 ). The positive predictive value for an individual to be a 2 + 0 carrier after testing positive for the g.27134T>G SNP with two copies of SMN1 is highest among Ashkenazi Jews (~100%) but lower in other ethnic groups, ranging from 1 in 99 to 1 in 39 ( Table 3 ).
SMN1 sequence pathogenic variants identified by NGS
Among all samples analyzed for sequence variants by NGS, we identified 10 individuals with potentially pathogenic single-nucleotide variants in the SMN1 gene. These variants were either previously found in patients with SMA or novel likely pathogenic variants (Supplementary Table S3 online). We confirmed the NGS results using gene-specific PCR followed by amplicon-based sequencing (Supplementary Figure S5 online, Supplementary Methods and Procedures online).
Discussion
NGS has enabled tremendous progress in clinical molecular testing, including population-based expanded carrier screening.15,16,31 A recent large cohort study suggested that an expanded carrier screen involving NGS increases detection rates for a variety of potentially serious genetic diseases when compared with current recommendations, which focus on testing a small number of diseases in high-risk populations.31 While NGS generates reliable SNV results in a high-throughput mode and can be used for CNV analysis, calling sequence variants and CNVs for genes with highly homologous sequences is technically challenging. For this reason, SMN1 and SMN2 have been put into a “dead zone” of genes that are not amenable to accurate NGS alignment.19
The majority of SMN1 and SMN2 NGS short reads lack informative PSVs for accurate mapping, and simple depth-of-coverage analyses cannot be used directly for gene-specific copy-number analysis. However, ambiguously aligned reads (i.e., reads aligned to SMN1 or SMN2) may be used to calculate the total combined copy number of SMN1 and SMN2. Gene-specific reads containing the c.840C/T PSV can then be used to calculate the SMN1 to SMN2 copy-number ratio and in turn permit derivation of gene-specific copy number. We used this approach to analyze 6,738 samples submitted to our laboratory for carrier testing. Measures of test reproducibility, sensitivity, and specificity indicate that this NGS method is highly accurate and robust for SMN1 copy-number analysis.
A recent study identified several SNPs, including g.27134T>G, that are tightly linked to a haplotype in 2 + 0 carriers who have two copies of SMN1 in tandem duplication on one chromosome and zero copies on the other.22 Since our carrier screening panel was designed to analyze the entire coding sequence and flanking intronic regions of every gene on the panel, including SMN1 and SMN2, we were able to detect clinically relevant SMN1 sequence variants (e.g., g.27134T>G) in addition to copy-number changes. We determined SMN1 copy number and genotyped the g.27134T>G SNP in different ethnic groups and found that this approach increases SMA carrier detection rates in all ethnic groups compared with conventional methodologies. The positive predictive value for an individual to be a SMA carrier when SMN1 copy number is two and the g.27134T>G SNP is present is highest for Ashkenazi Jews (~100%), which is consistent with a previous study.22 The positive predictive value was much lower for the general Asian population (~1.6%), however, in contrast to the same previous report (~100%). This discrepancy could be due to sampling differences, as distinct Asian subpopulations were included in our study ( Table 3 ). It should be noted that only a fraction of SMN1 duplicated alleles were linked to the g.27134T>G SNP in individuals other than African Americans, and further study is necessary to identify haplotypes linked to duplication alleles in these populations. Finally, we were able to identify pathogenic or likely pathogenic SMN1 single-nucleotide variants in 10 individuals, consistent with an overall carrier frequency of 0.15% in our cohort.
In summary, our NGS test reported herein is a sensitive and robust assay of SMN1 copy-number and sequence variation that increases SMA carrier detection rates across all populations. This approach can be integrated into existing NGS-based carrier screening panels to improve SMA detection rates and reduce the overall cost of population carrier screening.
Disclosure
Y.F., X.G., L.M., J.S., J.L., X.T., T.Z., W.J., H.C., X.W., M.T., P.L., H.M., Y.W., F.L., E.S.S., W.V.Z., D.M., S.W., Z.C., Y.Y., A.L.B., C.M.E., F.X., L.J.W., and J.Z. are faculty members or employees in the Joint Venture of Baylor Genetics Laboratories and Baylor College of Medicine. The Baylor Genetics Laboratories offer extensive fee-based genetic tests including the use of massively parallel sequencing for carrier screening. The other authors declare no conflict of interest.
References
Emery AE, Hausmanowa-Petrusewicz I, Davie AM, Holloway S, Skinner R, Borkowska J. International collaborative study of the spinal muscular atrophies. Part 1. Analysis of clinical and laboratory data. J Neurol Sci 1976;29:83–94.
Dubowitz V. Chaos in the classification of SMA: a possible resolution. Neuromuscul Disord 1995;5:3–5.
Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol 2005;57:704–712.
Hendrickson BC, Donohoe C, Akmaev VR, et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J Med Genet 2009;46:641–644.
Prior TW ; Professional Practice and Guidelines Committee. Carrier screening for spinal muscular atrophy. Genet Med 2008;10:840–842.
MacDonald WK, Hamilton D, Kuhle S. SMA carrier testing: a meta-analysis of differences in test performance by ethnic group. Prenat Diagn 2014;34:1219–1226.
Feldkötter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002;70:358–368.
Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA 1999;96:6307–6311.
Lindsay SJ, Khajavi M, Lupski JR, Hurles ME. A chromosomal rearrangement hotspot can be identified from population genetic variation and is coincident with a hotspot for allelic recombination. Am J Hum Genet 2006;79:890–902.
van der Steege G, Grootscholten PM, van der Vlies P, et al. PCR-based DNA test to confirm clinical diagnosis of autosomal recessive spinal muscular atrophy. Lancet 1995;345:985–986.
McAndrew PE, Parsons DW, Simard LR, et al. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet 1997;60:1411–1422.
Cuscó I, Barceló MJ, Baiget M, Tizzano EF. Implementation of SMA carrier testing in genetic laboratories: comparison of two methods for quantifying the SMN1 gene. Hum Mutat 2002;20:452–459.
Arkblad EL, Darin N, Berg K, et al. Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscul Disord 2006;16:830–838.
Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014;312:1870–1879.
Hallam S, Nelson H, Greger V, et al. Validation for clinical use of, and initial clinical experience with, a novel approach to population-based carrier screening using high-throughput, next-generation DNA sequencing. J Mol Diagn 2014;16:180–189.
Abulí A, Boada M, Rodríguez-Santiago B, et al. NGS-based assay for the identification of individuals carrying recessive genetic mutations in reproductive medicine. Hum Mutat 2016;37:516–523.
Retterer K, Scuffins J, Schmidt D, et al. Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genet Med 2015;17:623–629.
Feng Y, Chen D, Wang GL, Zhang VW, Wong LJ. Improved molecular diagnosis by the detection of exonic deletions with target gene capture and deep sequencing. Genet Med 2015;17:99–107.
Mandelker D, Schmidt RJ, Ankala A, et al. Navigating highly homologous genes in a molecular diagnostic setting: a resource for clinical next-generation sequencing. Genet Med 2016;18:1282–1289.
Cuscó I, Barceló MJ, del Rio E, et al. Characterisation of SMN hybrid genes in Spanish SMA patients: de novo, homozygous and compound heterozygous cases. Hum Genet 2001;108:222–229.
Larson JL, Silver AJ, Chan D, Borroto C, Spurrier B, Silver LM. Validation of a high resolution NGS method for detecting spinal muscular atrophy carriers among phase 3 participants in the 1000 Genomes Project. BMC Med Genet 2015;16:100.
Luo M, Liu L, Peter I, et al. An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy. Genet Med 2014;16:149–156.
Prior TW, Nagan N, Sugarman EA, et al. Addendum to “Technical standards and guidelines for spinal muscular atrophy testing.” Genet Med 2016;18:752.
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002;30:e57.
Forreryd A, Johansson H, Albrekt AS, Lindstedt M. Evaluation of high throughput gene expression platforms using a genomic biomarker signature for prediction of skin sensitization. BMC Genomics 2014;15:379.
Liu CG, Calin GA, Meloon B, et al. An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc Natl Acad Sci USA 2004;101:9740–9744.
Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502–1511.
Sugarman EA, Nagan N, Zhu H, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet 2012;20:27–32.
Contreras-Capetillo SN, Blanco HL, Cerda-Flores RM, et al. Frequency of SMN1 deletion carriers in a Mestizo population of central and northeastern Mexico: A pilot study. Exp Ther Med 2015;9:2053–2058.
Sheng-Yuan Z, Xiong F, Chen YJ, et al. Molecular characterization of SMN copy number derived from carrier screening and from core families with SMA in a Chinese population. Eur J Hum Genet 2010;18:978–984.
Haque IS, Lazarin GA, Kang HP, Evans EA, Goldberg JD, Wapner RJ. Modeled fetal risk of genetic diseases identified by expanded carrier screening. JAMA 2016;316:734–742.
Author information
Authors and Affiliations
Supplementary information
Supplementary Information
(ZIP 1631 kb)
Rights and permissions
About this article

Cite this article
Feng, Y., Ge, X., Meng, L. et al. The next generation of population-based spinal muscular atrophy carrier screening: comprehensive pan-ethnic SMN1 copy-number and sequence variant analysis by massively parallel sequencing. Genet Med 19, 936–944 (2017). https://doi.org/10.1038/gim.2016.215
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2016.215
Keywords
This article is cited by
-
Screening and prenatal diagnosis of survival motor neuron gene deletion in pregnant women in Zhaoqing city, Guangdong Province
BMC Medical Genomics (2023)
-
Population WGS-based spinal muscular atrophy carrier screening in a cohort of 1076 healthy Polish individuals
Journal of Applied Genetics (2023)
-
Next generation sequencing is a highly reliable method to analyze exon 7 deletion of survival motor neuron 1 (SMN1) gene
Scientific Reports (2022)
-
Genetic deconvolution of fetal and maternal cell-free DNA in maternal plasma enables next-generation non-invasive prenatal screening
Cell Discovery (2022)
-
NGS-based spinal muscular atrophy carrier screening of 10,585 diverse couples in China: a pan-ethnic study
European Journal of Human Genetics (2021)
