
Abstract
Disclaimer: These recommendations are designed primarily as an educational resource for medical geneticists and other healthcare providers to help them provide quality medical services. Adherence to these recommendations is completely voluntary and does not necessarily assure a successful medical outcome. These recommendations should not be considered inclusive of all proper procedures and tests or exclusive of other procedures and tests that are reasonably directed toward obtaining the same results. In determining the propriety of any specific procedure or test, the clinician should apply his or her own professional judgment to the specific clinical circumstances presented by the individual patient or specimen. Clinicians are encouraged to document the reasons for the use of a particular procedure or test, whether or not it is in conformance with this statement. Clinicians also are advised to take notice of the date this statement was adopted and to consider other medical and scientific information that becomes available after that date. It also would be prudent to consider whether intellectual property interests may restrict the performance of certain tests and other procedures.
To promote standardized reporting of actionable information from clinical genomic sequencing, in 2013, the American College of Medical Genetics and Genomics (ACMG) published a minimum list of genes to be reported as incidental or secondary findings. The goal was to identify and manage risks for selected highly penetrant genetic disorders through established interventions aimed at preventing or significantly reducing morbidity and mortality. The ACMG subsequently established the Secondary Findings Maintenance Working Group to develop a process for curating and updating the list over time. We describe here the new process for accepting and evaluating nominations for updates to the secondary findings list. We also report outcomes from six nominations received in the initial 15 months after the process was implemented. Applying the new process while upholding the core principles of the original policy statement resulted in the addition of four genes and removal of one gene; one gene did not meet criteria for inclusion. The updated secondary findings minimum list includes 59 medically actionable genes recommended for return in clinical genomic sequencing. We discuss future areas of focus, encourage continued input from the medical community, and call for research on the impact of returning genomic secondary findings.
Genet Med 19 2, 249–255.
Similar content being viewed by others

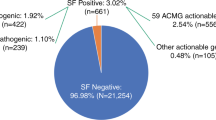
Background
The intent of the original incidental findings recommendations1 was that clinical diagnostic laboratories performing exome or genome sequencing should report known pathogenic (KP) or expected pathogenic (EP) variants in the 56 American College of Medical Genetics and Genomics (ACMG) genes even when unrelated to the primary medical reason for testing. Subsequently, the ACMG revised the terminology to “secondary findings” (SFs) because these genes are intentionally being analyzed, as opposed to genetic variants found incidentally or accidentally; the shift in terminology also maintained consistency with a recommendation by the Presidential Commission on Bioethical Issues.2 An additional modification to the original policy included offering an option to opt-out of receiving SFs for individuals undergoing clinical genomic sequencing (GS).3 This revision was due, in part, to results from a survey of ACMG members in which more than 80% of respondents supported an option for patients undergoing GS to decline SF analysis following appropriate counseling. In addition, more than 90% of respondents supported a minimum list of SFs that would be updated and refined over time. This article concluded that according to its membership, the ACMG “should update a list of genes to be assessed when clinical genome-scale sequencing is performed. Informed consent is necessary, and reporting of secondary findings should be optional.”4
The ACMG Board of Directors created the ACMG Secondary Findings Maintenance Working Group (SFWG) in 2014 to define and implement a process for updating the SF list. In March 2015, the process for nominating revisions to the list was announced to the ACMG community.
Materials and Methods
Nominations for genes/conditions to add to or remove from the SF list were accepted from ACMG members via a nomination form (see Supplementary file S1 online) that was developed through a subcommittee of the SFWG. The form was refined based on feedback from SFWG members and clinical genetics/genomics colleagues. Data collected included phenotypes associated with the gene, ordered by prevalence, and reported variants in the gene from ClinVar and the Human Gene Mutation Database. Also collected was information required to determine whether the gene/condition warranted inclusion based on clinical features, likelihood of early clinical diagnosis by a pediatrician/internist, molecular genetic characteristics, clinical genetic testing options, and medical actionability. Initially, medical actionability was evaluated according to a semiquantitative metric that included the following major adjudication criteria: severity of disease/nature of the health threat; likelihood of the disease/health threat materializing (i.e., penetrance); efficacy of specific intervention(s); and overall strength of the current knowledge base about the gene/condition. In July 2015, a fifth criterion was added: acceptability of the proposed intervention based on its risks and benefits. The SFWG acknowledged the inherent subjectivity and difficulty of rating any given intervention as it applies to an individual but voted unanimously in favor of adding this fifth criterion. This achieved consistency with efforts of the NIH-funded Clinical Genome Resource (ClinGen)5 Actionability Working Group,6 which had already applied these five criteria to assign actionability scores (called “Actionability Evidence-based Summaries”)7 to genes/conditions on the SF list.
Each nomination form was completed by an ACMG member or completed by a non-member and then submitted by an ACMG member. Completed forms were reviewed by the SFWG in a study section–like model. We also reviewed the Actionability Evidence-based Summary for any genes already reviewed by the ClinGen Actionability Working Group. Each form was presented in a SFWG meeting or conference call by the submitter or a designee. After thorough discussion of the rationale for making the proposed change, SFWG members voted on whether to accept the submitter’s recommendation.
Nominations that were recommended by the SFWG were sent to the ACMG Board of Directors with a summary of the SFWG discussion, voting outcome, and a recommendation for the suggested change to the SF minimum list. The Board reviewed each recommendation on a gene-by-gene basis in July 2016 and September 2016.
The SFWG discussed and unanimously agreed on a nomenclature to support versioning of the SF list, reflecting both major and minor revisions. Major revisions could be necessary due to conceptual changes to the categories of genes/variants included or the addition or removal of many genes in a single update. A major revision will be denoted by changing the version number to the next integer (v3.0, v4.0, etc.). Minor revisions reflect addition or removal of one or a few genes or variants and will be denoted by changing the number after the decimal point (v2.1, v2.2, etc.). Any new version number will be followed by the month and year in which the update was published. The ACMG Board has approved this versioning nomenclature.
Results
To refine the nomination form to collect all relevant information, SFWG members and colleagues were requested to complete nomination forms for 28 genes already on the SF list. Of these, the SFWG unanimously supported removal of one gene: MYLK associated with familial thoracic aortic aneurysm and dissection (FTAAD).
Between March 2015 and May 2016, five nominations for additions to the SF list were received and evaluated by the SFWG. One of these, PTCH1 associated with Gorlin syndrome/nevoid basal cell carcinoma syndrome, did not achieve SFWG consensus for addition due to insufficient evidence that knowledge of a KP/EP variant in the gene would alter medical management. Each of the other four genes—ATP7B associated with Wilson disease, BMPR1A and SMAD4 associated with juvenile polyposis, and OTC associated with ornithine transcarbamylase deficiency—received a unanimous vote from SFWG members for addition to the list. The ACMG Board subsequently reviewed and approved each of the five recommendations of the SFWG: removal of MYLK and addition of ATP7B, BMPR1A, SMAD4, and OTC.
The following paragraphs describe these genes and the rationale for removing or adding each of them according to the criteria outlined above.
Key features of MYLK include the rarity of KP variants, all of which are loss-of-function variants, as well as the lack of an effective confirmatory test, screening modality, or intervention to prevent or reduce morbidity or mortality from FTAAD. Because aortic dissection related to variants in MYLK often presents without a history of aortic enlargement, the intervention of echocardiogram may be falsely reassuring and may not prevent sudden cardiac death.8 In addition, there is insufficient evidence on the appropriate age to begin medications and the efficacy of this intervention to reduce dynamic stress on the aorta.9 The SFWG considered that additional data on MYLK are likely to be gathered in the coming years from aneurysm genetic testing panels, and this evidence could be re-evaluated periodically for possible re-addition of MYLK to the SF list.
ATP7B is associated with autosomal-recessive Wilson disease (MIM 277900). Morbidity among homozygotes directly correlates with copper deposition in the liver, brain, and eye. The disease is progressive, and, if left untreated, premature death is likely. In some cases, liver failure may be the presenting sign. Given its long recognition as a Mendelian disorder, it is reasonable to consider Wilson disease to be at least relatively highly penetrant. Expressivity is variable. Treatment for Wilson disease involves administration of copper chelating agents and/or zinc to block intestinal absorption of copper; treatment is extremely effective when administered prior to the onset of symptoms.10,11,12 Sanger sequencing of the ATP7B gene is considered confirmatory in asymptomatic patients. In symptomatic patients, in addition to Sanger sequencing, the results of serum ceruloplasmin, serum copper concentration, and 14-hour urine copper excretion can be diagnostic. The ClinGen Actionability scoring process generated a high actionability score of 10/12 for copper chelation and zinc therapy in the treatment of ATP7B-associated liver disease and/or neuropsychiatric disease.7 Based on this evidence, the SFWG voted unanimously to add ATP7B to the SF list for the recessive state in which two KP/EP variants are detected in trans through GS.
BMPR1A and SMAD4 are two genes known to cause autosomal-dominant juvenile polyposis syndrome (MIM 174900), which is associated with gastrointestinal (GI) bleeding, anemia, and increased risk of colon cancer and other GI cancers. An individual with a KP/EP variant in BMPR1A has a more than 90% chance of developing polyps and a 10–50% lifetime risk of developing cancer. The majority of patients with clinical features of the disease have colon polyps by age 20, and colon cancer typically develops by the mid-40s. Screening via endoscopy with histological confirmation of juvenile-type polyps is readily available, highly sensitive, effective at reducing morbidity, and presents minimal risk or burden. Although most individuals with a BMPR1A variant have a positive family history, many cases remain undetected because GI cancer screening is not routine in the general population before age 50. Penetrance of KP/EP variants in SMAD4 is estimated to be approximately 20% for colon cancer by age 35 and nearly 70% by age 65, with the average age of onset reported to be in the early 40s. Gastric cancer occurs in approximately 21% of patients with gastric polyps. Screening via colonoscopy and upper GI endoscopy for polyps are effective interventions for risk reduction.13 The SFWG unanimously agreed that juvenile polyposis syndrome associated with KP/EP variants in BMPR1A and SMAD4 meets criteria for addition to the SF list based on the existing knowledge base.
Physicians should be aware that hereditary hemorrhagic telangiectasia syndrome is another phenotype associated with loss-of-function variants in SMAD4. Hemorrhagic telangiectasia syndrome involves a risk of sudden death due to arteriovenous malformation rupture and aortopathy as well as morbidity due to anemia and/or hypoxia. Screening includes imaging and embolization for arteriovenous malformations.14 A patient with a KP/EP variant identified in SMAD4 is potentially at risk for juvenile polyposis and/or hemorrhagic telangiectasia syndrome and therefore should be screened for both diseases.
Ornithine transcarbamylase deficiency (MIM 311250) is a urea cycle defect caused by variants in the X-linked OTC gene. Severely affected males present within the first week of life with hyperammonemic coma that is associated with high neonatal mortality; long-term survivors have significant neurologic deficits and intellectual disability.15 Penetrance in hemizygous males is nearly 100%, although cases in which there is partial expression of the mutated allele with residual enzyme activity often present later with milder symptoms. In heterozygous females, penetrance estimates range from approximately 20 to 30%.16,17 The phenotype among affected females varies widely from mild to severe, depending primarily on the pattern of X-inactivation of the gene in hepatocytes.15 Interventions exist to prevent crises and ameliorate outcomes, including protein restriction in the diet and medications that remove ammonia by alternate metabolic pathways.18 These treatments are most useful in symptomatic heterozygous females and among males with later onset or partial disease. Medical management may also include the potential for prolonged hospitalization during illness as well as dialysis for acute crises with hyperammonemia to help prevent or minimize brain damage. For males with severe disease who present in the neonatal period, and for some males and females with later-onset disease, liver transplantation is recommended due to the high risk for recurrent hyperammonemia, subsequent neurological deterioration, and/or death.19 OTC deficiency is included on newborn screening panels in only some states; however, males with severe disease often become ill in the first 2–3 days of life, when newborn screening results are not yet available. Given the morbidity and mortality risks, the SFWG voted unanimously to add ornithine transcarbamylase deficiency associated with KP/EP variants in the OTC gene to the SF list.
In selecting the variant types to recommend for return, the SFWG considered using the terminology of “pathogenic” and “likely pathogenic” according to updated variant classification criteria20 published since the original policy statement, but ultimately decided to maintain the terminology of KP and EP. This decision was based on a recognition that classifying a variant as likely pathogenic is more burdensome for laboratories, whereas an EP classification is simpler because it is limited to loss-of-function variants and novel missense variants need not be analyzed as SFs.
Table 1 depicts the “ACMG SF v2.0 (September, 2016)” updated list of SFs recommended for return from clinical GS. Table 2 summarizes the updates to the SF list.
Discussion
This article highlights the process for nominating changes to the ACMG Secondary Findings minimum list. This process and form were piloted by SFWG members and colleagues before soliciting nominations from the ACMG membership. Goals for the nomination process were to (i) make the nomination form accessible through the ACMG website; (ii) collect information from the broader community to inform decision-making by the SFWG and the Board; and (iii) update the gene list at least annually in a way that is inclusive of ACMG members. We hope to transition to a web-based nomination process such that submission of supporting information would directly populate a database.
The nomination form has been designed to accept nominations for additions to as well as removals from the SF list. Five formal nominations for adding genes/conditions to the SF list were received between March 2015 and March 2016, and four of these have been added to the ACMG SF v2.0 list based on unanimous consensus among SFWG members and approval by the ACMG Board. In addition, one gene has been recommended and approved for removal. It is our hope that this article will stimulate additional nominations from the ACMG membership and broader medical community for genes to consider adding to the ACMG SF v2.0 list, as well as further assessment of the evidence to support genes/conditions already on the list.
Our group has attempted to uphold the principles outlined in the original policy statement1 while expanding on certain aspects. We have continued to focus primarily on SFs related to monogenic disorders for which there is evidence of clinical utility. Additionally, we have attempted to standardize the evaluation of current and prospective genes by adopting a process that includes a semiquantitative metric for determining actionability that is consistent with the goals of the ClinGen Actionability Working Group. We continue to support the reporting of known or expected pathogenic variants, but we do not recommend reporting variants of uncertain significance as SFs. Among the challenges inherent in developing and curating this list, we recognize the presumption of high penetrance for these genes and diseases based on potentially biased case ascertainment. Specifically, literature reports for many of these conditions represent assessment of disease probands and their families. Over time, we may discover that penetrance for some of these conditions is lower than current estimates suggest. We anticipate that the increasing availability of large population databases, such as the Exome Aggregation Consortium (ExAC), will be helpful in refining estimates of penetrance, which may inform future curation of the ACMG SF v2.0 list.
Future Focus of the Secondary Findings Maintenance Working Group
Considering pharmacogenomic variants
Although the main mission of the SFWG is to focus on genetic changes with high penetrance for actionable phenotypes, incorporation of pharmacogenomic (PGx) variants onto a SF minimum list has also been considered. A preliminary version of a PGx nomination form is being developed, and SFWG discussions have focused on the actionability of findings for PGx variants. Guidelines from the Clinical Pharmacogenetics Implementation Consortium provide specific medical management recommendations on the basis of PGx results; however, assessing the impact of these results on clinical outcomes has proven difficult due to a lack of randomized, controlled trials and small numbers of patients for whom PGx results are available.
Implementation of PGx results based on secondary findings presents additional challenges. Calling sequence variants can be complex for several reasons: (i) in some cases, the presence of pseudogenes may confound sequence results; (ii) certain actionable PGx “variants” are actually haplotypes composed of several variants, some of which are intronic and not covered by standard WES; (iii) the “star allele” nomenclature used for PGx variants is not familiar to most geneticists and needs simplification; and (iv) each testing laboratory would need to be provided with specific genomic coordinates to analyze to generate PGx diplotypes. There are also challenges related to implementation of PGx results in clinical practice. Results from single gene disorders may require action in the near term, such as initiation of surveillance, whereas PGx results are not actionable until the decision is made to prescribe a drug related to the PGx variant. The prescription of a drug at that time arguably offers a window for genetic testing for variants proven to be important in the drug’s administration; thus, proactive assessment may not be necessary. Finally, the need for certain drugs is rare and may never arise for an individual. However, some rare drugs have such severe side effects, including death, that identification of PGx variants is critical. For these reasons, we are focusing our initial attention on coding variants related to commonly prescribed medications as well as medications associated with serious adverse events for which there is greater urgency surrounding actionability.
Expanding the nomination process
The SFWG plans to accept nominations from other professional specialty organizations, such as professional groups of genetic counselors, primary care clinicians, and other clinical specialists who may order or return results from GS and thereby encounter SFs in their practice. We hope that this process will help to engage the broader medical community in considering the implications of genomic SFs. The ACMG also intends to develop resources to assist clinicians in medical management based on specific SFs.
Assessing the impact of secondary findings
We recognize that the potential impact of reporting actionable SFs is significant and far-reaching. The SFWG hopes this process of nominations and updates will serve as a reference point in related efforts, such as initiatives that apply genetic and genomic testing in public health programs and population screening. Successful efforts have already been directed toward evaluating the efficacy of newborn screening panels in detecting genetic diseases,21,22 and a similar approach could begin to evaluate the utility of certain applications of genomic screening in healthy adults. The reporting of SFs presents significant opportunities to prevent disease, but this process may introduce fiscal and other costs, including those associated with further confirmatory testing. As clinical GS becomes less expensive and more widespread, the amount of data and time required to communicate this information to patients will likely increase, as will the need for genetics/genomics education among primary care clinicians. The medical, legal, social, and economic implications of these trends are broad, and it will be important for the genomics community to study the impact of SFs on the busy clinician, individual patients and families, and populations at large.
Disclosure
S.S.K. is a consultant to Genome Medical and Helix. S.J.B. is an employee and stockholder in OPKO, of which GeneDx is a subsidiary. W.K.C. is a member of the scientific advisory board of Regeneron Genetic Center. C.E. provides services to Baylor Miraca Genetics Laboratories through a professional services agreement as Chief Medical Officer and Chief Quality Officer. G.E.H. participated in a roundtable discussion sponsored by 23andMe. T.E.K. is a paid scientific advisor for Rxight Pharmacogenetics. B.R.K. has provided consultation to Novartis, Alexion, and AstraZeneca and serves on the Medical Advisory Board of Accolade. He has prepared a CME educational program sponsored by Axis, spoke at a meeting sponsored by Sequenom, and participated in a roundtable discussion sponsored by 23andMe. He is a founding member of Envision Genomics and serves on the board of directors of American College of Medical Genetics and Genomics Foundation and Children’s Tumor Foundation. C.L.M. is a consultant to The Jackson Laboratory. D.T.M. is a part-time clinical consultant to Claritas Genomics (nonequity professional services agreement). The other authors declare no conflict of interest.
References
Green RC, Berg JS, Grody WW, et al.; American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013;15:565–574.
Weiner C. Anticipate and communicate: ethical management of incidental and secondary findings in the clinical, research, and direct-to-consumer contexts (December 2013 report of the Presidential Commission for the Study of Bioethical Issues). Am J Epidemiol 2014;180:562–564.
ACMG Board of Directors. ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing. Genet Med 2015;17:68–69.
Scheuner MT, Peredo J, Benkendorf J, et al. Reporting genomic secondary findings: ACMG members weigh in. Genet Med 2015;17:27–35.
Rehm HL, Berg JS, Brooks LD, et al.; ClinGen. ClinGen–the Clinical Genome Resource. N Engl J Med 2015;372:2235–2242.
Hunter JE IS, Biesecker LG, Buchanan A, et al. A standardized, evidence-based protocol to assess clinical actionability of genetic disorders associated with genomic variation. Genet Med. 2016;18:467–475.
ClinGen. Actionability Working Group Evidence-based Summaries. https://clinicalgenome.org/working-groups/actionability/projects-initiatives/actionability-evidence-based-summaries. Accessed 23 August, 2016.
Wang L, Guo DC, Cao J, et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet 2010;87:701–707.
Milewicz DM, Regalado RE. Thoracic Aortic Aneurysms and Aortic Dissections. GeneReviews. University of Washington: Seattle, WA, 2012.
Weiss KH. Wilson Disease. GeneReviews. University of Washington: Seattle, WA, 2016.
Roberts EA, Schilsky ML ; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008;47:2089–2111.
Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson’s disease. Lancet 2007;369:397–408.
Larsen Haidle J, Howe JR. Juvenile Polyposis Syndrome. GeneReviews. University of Washington, Seattle, WA, 2015.
McDonald J, Pyeritz RE. Hereditary Hemorrhagic Telangiectasia. GeneReviews. University of Washington, Seattle, WA, 2014.
Tuchman M. The clinical, biochemical, and molecular spectrum of ornithine transcarbamylase deficiency. J Lab Clin Med 1992;120:836–850.
Batshaw ML, Msall M, Beaudet AL, Trojak J. Risk of serious illness in heterozygotes for ornithine transcarbamylase deficiency. J Pediatr 1986;108:236–241.
Gyato K, Wray J, Huang ZJ, Yudkoff M, Batshaw ML. Metabolic and neuropsychological phenotype in women heterozygous for ornithine transcarbamylase deficiency. Ann Neurol 2004;55:80–86.
Batshaw ML, MacArthur RB, Tuchman M. Alternative pathway therapy for urea cycle disorders: twenty years later. J Pediatr 2001;138(1 suppl):S46–54; discussion S54.
Lichter-Konecki U, Caldovic L, Morizono H, et al. Ornithine Transcarbamylase Deficiency. GeneReviews. University of Washington, Seattle, WA, 2016.
Richards S, Aziz N, Bale S, et al.; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424.
Bodian DL, Klein E, Iyer RK, et al. Utility of whole-genome sequencing for detection of newborn screening disorders in a population cohort of 1,696 neonates. Genet Med 2016;18:221–230.
Berg JS, Powell CM. Potential uses and inherent challenges of using genome-scale sequencing to augment current newborn screening. Cold Spring Harb Perspect Med. 2015;5:pii: a023150.
Acknowledgements
We acknowledge the contributions of the original ACMG Incidental Findings Working Group for their groundbreaking efforts to begin the important process of identifying and reporting actionable genetic variants based on genome-scale sequencing, including those who are not members of the current SFWG: Robert C. Green, Jonathan S. Berg, Wayne W. Grody, Amy L. McGuire, Julianne M. O’Daniel, Heidi L. Rehm, Marc S. Williams, and Leslie G. Biesecker. We thank former members of the Secondary Findings Maintenance Working Group for their insights and feedback, specifically Robert Nussbaum, Will Parsons, and Richard Sharp. We are also grateful to cardiogenetics counselors at GeneDx who completed nomination forms for genes on the SF list to pilot this process; Kyle Retterer, manager of Exome BioInformatics at GeneDx, who provided exome data related to PGx variants; Michelle Carillo at CPIC, who prepared PGx summaries for SFWG review; and Katrina Goddard and the ClinGen Actionability Working Group for sharing their approach and evaluations of certain genes.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Supplementary information
Supplementary File
(PDF 233 kb)
Rights and permissions
About this article

Cite this article
Kalia, S., Adelman, K., Bale, S. et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 19, 249–255 (2017). https://doi.org/10.1038/gim.2016.190
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2016.190
Keywords
This article is cited by
-
Current prospects of hereditary adrenal tumors: towards better clinical management
Hereditary Cancer in Clinical Practice (2024)
-
Genomic susceptibility to gastric cancer in Northwest Iran: population-based and case–control studies
Egyptian Journal of Medical Human Genetics (2024)
-
A framework for the evaluation and reporting of incidental findings in clinical genomic testing
European Journal of Human Genetics (2024)
-
Parents’ attitudes towards research involving genome sequencing of their healthy children: a qualitative study
European Journal of Human Genetics (2024)
-
Islamic Viewpoints on Opportunistic Sex Selection of IVF Embryos upon doing Preimplantation Genetic Testing for Preventing Genetic Diseases
Asian Bioethics Review (2024)
