
Abstract
Next-generation sequencing technologies have been and continue to be deployed in clinical laboratories, enabling rapid transformations in genomic medicine. These technologies have reduced the cost of large-scale sequencing by several orders of magnitude, and continuous advances are being made. It is now feasible to analyze an individual’s near-complete exome or genome to assist in the diagnosis of a wide array of clinical scenarios. Next-generation sequencing technologies are also facilitating further advances in therapeutic decision making and disease prediction for at-risk patients. However, with rapid advances come additional challenges involving the clinical validation and use of these constantly evolving technologies and platforms in clinical laboratories. To assist clinical laboratories with the validation of next-generation sequencing methods and platforms, the ongoing monitoring of next-generation sequencing testing to ensure quality results, and the interpretation and reporting of variants found using these technologies, the American College of Medical Genetics and Genomics has developed the following professional standards and guidelines.
Genet Med 15 9, 733–747.
Similar content being viewed by others


A. Introduction
Sequencing technologies have evolved rapidly over the past 5 years. Semi-automated Sanger sequencing has been used in clinical testing for many years and is still considered the gold standard. However, its limitations include low throughput and high cost, making multigene panels laborious and expensive. Recent technological advancements have radically changed the landscape of medical sequencing. Next-generation sequencing (NGS) technologies utilize clonally amplified or single-molecule templates, which are then sequenced in a massively parallel fashion. This increases throughput by several orders of magnitude. NGS technologies are now being widely adopted in clinical settings. Three main levels of analysis, with increasing degrees of complexity, can now be performed via NGS: disease-targeted gene panels, exome sequencing (ES), and genome sequencing (GS). All have advantages over Sanger sequencing in their ability to sequence massive amounts of DNA, yet each also has challenges for clinical testing.
A.1. Disease-targeted gene panels
Disease-targeted gene panels interrogate known disease-associated genes. Focusing on a limited set of genes allows greater depth of coverage for increased analytical sensitivity and specificity. Greater depth of coverage increases the confidence in heterozygous calls and the likelihood of detecting mosaicism or low-level heterogeneity in mitochondrial or oncology applications. Furthermore, because only genes with an established role in the targeted disease are sequenced, the ability to interpret the findings in a clinical context is greater. Follow-up Sanger sequencing or an alternative technology can be used to fill gaps in the NGS data for regions showing low coverage (e.g., GC-rich or repetitive regions), which improves clinical sensitivity of the assay. Targeting fewer genes also allows the laboratory to use desktop sequencers and run more patient samples per instrument cycle (barcoding and pooling) as compared with ES/GS. The amount of data and storage requirements are also more manageable.
A.2. Exome sequencing
ES attempts to cover all coding regions of the genome. The exome is estimated to comprise ~1–2% of the genome, yet contains ~85% of recognized disease-causing mutations.1 Presequencing sample preparation is required to enrich the sample for the targeted coding regions. Current estimates of exome coverage through NGS are between 90 and 95%.2 At this time, enrichment is performed by in-solution hybridization methods. However, certain regions of the exome are still not amenable to this method of enrichment and NGS due to sequence complexity. ES is used for detecting variants in known disease-associated genes as well as for the discovery of novel gene–disease associations. Gene discovery has historically been limited to research laboratories. This is now changing with the ability to identify novel disease-gene candidates in the clinical laboratory, although further studies, often in collaboration with research laboratories, are required to prove the association. Coverage and cost of ES will be between those of targeted gene panels and GS. One strategy some clinical laboratories are adopting is to perform ES but proceed with the interpretation of only genes already known to be associated with disease. If no mutation that can explain the patient’s symptoms is identified, the data can be reanalyzed for the remaining exome to potentially identify new disease–gene associations. In a study from the National Institutes of Health for rare and ultrarare disorders, ES provided a diagnosis in nearly 20% of cases.3 However, because the depth of coverage for an exome is not uniform, the analytical sensitivity for ES may be lower than the sensitivity for most targeted gene panels, given that a substantial number of exons in known disease-associated genes may lack sufficient coverage to make a sequence call. Although Sanger sequencing is commonly used to fill in missing content in disease-targeted test panels, the scope of ES makes this strategy impractical, expensive, and rarely used. Analytical specificity may also be compromised with less depth of coverage, requiring more Sanger testing to prevent false-positive (FP) variant calls.
A.3. Genome sequencing
GS covers both coding and noncoding regions. One advantage of GS is that presequencing sample preparation is straightforward, not requiring PCR or hybridization enrichment strategies for targeted regions. Due to limitations in the interpretation of noncoding variants, coding regions are often analyzed initially. If causative mutations are not found, data can then be reanalyzed to look for regulatory variants in noncoding regions that may affect expression of disease-associated genes. Data can also be examined for copy-number variants (CNVs) or structural variants that may either be outside of the coding regions or more easily detected using GS due to increased quantitative accuracy. GS is currently the most costly technology with the least average depth of coverage, although these limitations are likely to diminish in the future.
This document describes the standards and guidelines for clinical laboratories performing NGS for the assessment of targeted gene panels, the exome, and the genome. Given the rapid pace with which this area of molecular diagnostics is advancing, this document attempts to cover issues essential for the development of any NGS test. It does not address specific technologies in detail and may not cover all issues relevant to each test application or characteristics unique to a specific platform. For example, this document does not focus on testing related to somatic variation and other mixed populations of cells, RNA applications of NGS, or the detection of circulating fetal DNA, and therefore additional considerations specific to these applications may be required.
B. Overview of NGS
NGS involves three major components: sample preparation, sequencing, and data analysis (as illustrated in Figure 1 ). The process begins with extraction of genomic DNA from a patient sample, and some approaches (e.g., targeted panels and ES) will include enrichment strategies to focus on a subset of genomic targets. A set of short DNA fragments (100–500 base pairs) flanked by platform-specific adapters is the required input for most currently available NGS platforms. A series of processing steps is necessary to convert the DNA sample into the appropriate format for sequencing. Multiple commercial sequencing platforms have been developed, all of which have the capacity to sequence millions of DNA fragments in parallel. Differences in the sequencing chemistry of each platform result in differences in total sequence capacity, sequence read length, sequence run time, and final quality and accuracy of the data. These characteristics may influence the choice of platform to be used for a specific clinical application. When the sequencing is complete, the resulting sequence reads are processed through a computational pipeline designed to detect DNA variants. Commonly used sample preparation methods, sequencing platforms, and steps in data analysis are briefly described below.

Next-generation sequencing involves three major components: sample preparation, sequencing, and data analysis.
B.1. Sample preparation
NGS may be performed on any sample type containing DNA, as long as the quality and quantity of the resulting DNA are sufficient. The laboratory should specify the required sample type and quantity based on their validation data. Given the complexity of the procedures and likelihood of manual steps, processes to prevent sample mix-up or to confirm final results must be employed as discussed in subsequent sections.
B.2. Library generation
Library generation is the process of creating random DNA fragments, of a certain size range, that contain adapter sequences on both ends. The adapters are complementary to platform-specific PCR and sequencing primers. Fragmentation of genomic DNA can be achieved through multiple methods, each having strengths and weaknesses. For most applications/platforms, PCR amplification of the library is necessary before sequencing.
B.3. Barcoding
Barcoding refers to the molecular tagging of samples with unique sequence-based codes, typically consisting of three or more base pairs. This enables pooling of patient samples, thereby reducing the per-sample processing cost. The number of samples that can be pooled will depend on the desired coverage of the region to be sequenced. Barcodes can be part of the adapter or can be added as part of a PCR enrichment step that is included in most protocols.
B.4. Target enrichment
Unless GS is performed, the genes or regions of interest must be isolated before sequencing. The targets can range from a relatively small number of genes (e.g., all genes associated with a specific disease) to the entire exome (all known protein-coding exons). Target enrichment approaches can be divided into multiplexed PCR-based methods (single or multiplex PCR or droplet PCR) and solid or in-solution oligonucleotide hybridization–based methods. Hybridization-based capture can be used for ES, but PCR-based approaches currently do not scale to that size. Strategies for target enrichment are reviewed in Mamanova et al.4
B.5. Sequencing platforms
Currently available commercial platforms are based on the ability to perform many parallel chemical reactions in a manner that allows the individual products to be analyzed. Chemistries include sequencing by synthesis or sequencing by ligation with reversible terminators, bead capture, and ion sensing.5 Each platform has specific parameters relevant to the laboratory and test requirements including instrument size, instrument cost, run time, read length, and cost per sample.
B.6. Data analysis
Given the huge amount of sequence data produced by NGS platforms, the development of accurate and efficient data handling and analysis pipelines is essential. This requires extensive bioinformatics support and hardware infrastructure. NGS data analysis can be divided into four primary operations: base calling, read alignment, variant calling, and variant annotation. Base calling is the identification of the specific nucleotide present at each position in a single sequencing read; this is typically integrated into the instrument software given the technology-specific nature of the process. Read alignment involves correctly positioning short DNA sequence reads (often 50–400 base pairs) along the genome in relation to a reference sequence. Variant calling is the detection of the DNA variants in the sequence analyzed as compared with a reference sequence. The accuracy of identifying variants greatly depends on the depth of sequence coverage; increased coverage improves variant calling. Because some regions may have low sequence coverage, it is important to track positions where there is absent data or an ambiguous call, enabling test limitations to be defined. Variant annotation adds information about each variant detected. For example, annotation pipelines will determine whether a variant is within or near a gene, where the variant is located within that gene (e.g., untranslated region, exon, intron), and whether the variant causes a change in an amino acid within the encoded protein. Ideally, the annotation will also include additional information that facilitates interpretation of its clinical significance. This information may include the presence of the variant in certain databases, the degree of evolutionary conservation of the encoded amino acid, and a prediction of whether the variant is pathogenic due to its potential impact on protein function using in silico algorithms.
C. Test Ordering
In the traditional genetic testing setting for disease diagnosis, the ordering physician’s role is to generate a differential diagnosis based on clinical features, family history, and physical examination. Genetic test(s) may be ordered to confirm or exclude a diagnosis. Laboratories perform the ordered genetic test(s) and report the presence of all potentially deleterious variants in the gene(s) analyzed as well as the methods and performance parameters of each test. Targeted NGS panels represent the logical extension of current sequencing tests for genetically heterogeneous disorders. By limiting the content of the test to just the regions relevant to a given disease, the resulting data usually have higher analytical sensitivity and specificity for detecting mutations. In addition, the laboratory is more likely to employ approaches to ensure complete coverage of the targeted regions such as performing Sanger sequencing for those areas with inadequate coverage. Finally, a more thorough analysis and interpretation of the data to account for all known variation previously associated with the disease may be performed. Targeted NGS panels may also conform more readily to current models of reimbursement for molecular diagnostic tests. Therefore, it may be more appropriate to initiate testing with disease-targeted panels before proceeding to ES or GS approaches until the technical and interpretative quality of ES/GS reaches that of disease-targeted testing.
By contrast, the use of ES or GS enables a broader hypothesis-free approach to testing the patient. Consequently, these tests require more collaboration between the laboratory and health-care providers to enable appropriate data interpretation. ES and GS approaches are currently being utilized primarily for cases in which disease-targeted testing is unavailable or was already conducted and did not explain the patient’s phenotype. However, this is likely to change in the future. Performing an ES or GS test but then initially restricting the analysis to a disease-associated set of genes based on the patient’s clinical indications may also be considered. If this is done, the laboratory needs to state the relevant parameters (e.g., coverage of the specific genes) in the patient’s report, allowing the physician to compare the performance of the ES/GS test to an available disease-specific panel test. Careful consideration for the potential impact of incidental findings, which may include carrier status for recessive diseases, must also be weighed given the context in which data are analyzed and returned to the patient. The laboratory director is responsible for describing both the advantages and limitations of their test offerings so that the health-care provider can make an informed decision.
Regardless of the approach employed, it is recommended that referring physicians provide detailed phenotypic information to assist the laboratory in analyzing and interpreting the results of testing. This step is necessary for ES and GS to enable appropriate filtering strategies to be employed. It is also highly recommended for large disease panel testing, given the diversity of genes and subphenotypes that may be included in a test panel. The ability for laboratories to prioritize variants for further consideration or likely relevance may be dependent on the constellation of existing symptoms and findings in the patient and future clinical evaluation in collaboration with the health-care providers. Furthermore, the ability to increase knowledge of variant significance is greatly aided by laboratories receiving and tracking patient phenotypes and correlating them with genotypes identified.
D. Upfront Considerations for Test Development
D.1. Test content
The factors relevant to test ordering described above also have an impact on the laboratory’s strategy for test development. Test development costs, analytical sensitivity and specificity, and analysis complexity are important factors that must be evaluated when considering development of NGS services.
D.1.1. Disease-targeted gene panels. It is recommended that the selection of genes and transcripts to be included in clinical disease-targeted gene panels using NGS be limited to those genes with sufficient scientific evidence for a causative role in the disease. Candidate genes without clear evidence of a disease association should not be included in these disease-targeted tests. If a disease-targeted panel contains genes for multiple overlapping phenotypes, laboratories should consider providing a physician the option of restricting the analysis to a subpanel of genes associated to the subphenotype (e.g., hypertrophic cardiomyopathy genes within a broad cardiomyopathy gene panel) to minimize the number of variants of unknown significance detected. Laboratories should also consider the expected number of detected variants and account for the time and expertise required for their evaluation. As a guide, the number of variants with potential clinical relevance will be approximately proportional to the size of the target region being analyzed.
D.1.2. Exome and genome sequencing. ES is a method of testing the RNA-coding exons and flanking splice sites of all recognized genes in the human genome. By contrast, GS attempts to sequence the entire three billion bases of the human genome. The ability to target the exome in ES is based on capture of the exome using a variety of reagent kits typically sold by commercial suppliers (see Clark et al.6 for a review). Laboratories should consider the differences between the commercially available reagents and be aware of refractory regions in each design. No exome capture method is fully efficient; therefore, laboratories must describe the method used and the capture efficiency expected based on their validation studies. Although GS is not dependent on capture limitations, not all regions of human DNA can be accurately sequenced with current methods (e.g., repetitive DNA), and therefore limitations of content, including limitations on assessing known disease-associated loci such as triplet repeat expansion diseases, must be described. Furthermore, if ES testing is marketed as a disease-targeted test, the coverage of high-contributing disease genes and variants, as determined during test validation, should be clearly noted in material accessible during test ordering and prominently noted on reports.
For targeted testing of diseases with significant genetic heterogeneity, ES/GS strategies may be more efficient, but limitations in gene inclusion and coverage must be clearly noted if such a test is marketed for a specific disease indication. If disease-associated variants are expected to occur beyond exonic regions (e.g., regulatory, copy-number, or structural variation), the test design strategy should include approaches to detect these types of variation. Laboratories may consider supplementing an ES assay with complementary assays to be able to detect all types of disease-associated variants. Alternatively, it must be clearly stated that these types of variants will not be detected. Inclusion of these variants may require supplementation of the standard assay or the use of companion technologies to improve the sensitivity of the test (e.g., adding capture probes designed for nonexonic regions to an ES test, Sanger sequencing to fill in low-coverage areas, molecular or cytogenetic methods to detect CNVs and structural variants).
D.2. Choice of sequencer and sequencing methods
In choosing a sequencer, the laboratory must carefully consider the size of the sequenced region, required depth of coverage, projected sample volume, turnaround time requirements, and costs. These considerations are important for a decision to invest in a desktop or stand-alone sequencer, which has a substantial difference in sequencing capacity. For example, long read lengths may be useful for certain applications, such as analysis of highly homologous regions. Short-read technologies may be sufficient for other applications. Because maximum read length is platform dependent, the test design, choice of platform, and choice of read length should be based on the type of variation that must be detected.
General sequencing modes include single-end sequencing (genomic DNA fragments are sequenced at one end only) and paired-end sequencing (both ends are sequenced). Paired-end sequencing increases the ability to map reads unambiguously, particularly in repetitive regions (see section D.5.) and has the added advantage of increasing coverage and stringency of the assay as bidirectional sequencing of each DNA fragment is performed. A variation of paired-end sequencing is mate-pair sequencing, which can be useful for structural variant detection.
Support for therapeutic decision making and targeted NGS gene panels in a prenatal setting requires quick turnaround as compared with other less time-sensitive applications. High-throughput instruments can reduce per-sample cost, but only if sufficient numbers of tests would be ordered. For disorders that require detection of variants below germline heterozygosity, such as somatic mutation testing in tumors and detection of heteroplasmic mitochondrial variants, approaches that achieve higher coverage should be taken. In addition, non-Sanger strategies to confirm low-level variation may be necessary, either through duplicate testing to increase confidence or through other complementary methods.
D.3. Choice of data analysis tools
D.3.1. Base calling. Each NGS platform has specific sequencing biases that affect the type and rates of errors made during the data-generation process. These can include signal-intensity decay over the read and erroneous insertions and deletions in homopolymeric stretches.7 Base-calling software that accounts for technology-specific biases can help address platform-specific issues. The best practice is to utilize a base-calling package that is designed to reduce specific platform-related errors. Generally, an appropriate, platform-specific base-calling algorithm is embedded within the sequencing instrument. Each base call is associated with a quality metric providing an evaluation of the certainty of the call. This is usually reported as a Phred-like score (although some software packages use a different quality metric and measure slightly different variables).
D.3.2. Read alignment. Various algorithms for aligning reads have been developed that differ in accuracy and processing speed. Depending on the types of variations expected, the laboratory should choose one or more read-alignment tools to be applied to the data. Several commercially available or open-source tools for read alignment are available that utilize a variety of alignment algorithms and may be more efficient for certain types of data than for others.8 Proper alignment can be challenging when the captured regions include homologous sequences but is improved by longer or paired-end reads. In addition, it is suggested that alignment to the full reference genome be performed, even for exome and disease-targeted testing, to reduce mismapping of reads from off-target capture, unless appropriate methods are used to ensure unique selection of targets.
D.3.3. Variant calling. The accuracy of variant calling depends on the depth of sequence coverage and improves with increasing coverage. The variant caller can differentiate between the presence of heterozygous and homozygous sequence variations on the basis of the fraction of reads with a given variant. It can also annotate the call with respect to proper genome and coding sequence nomenclature. Most variant-calling algorithms are capable of detecting single or multiple base variations, and different algorithms may have more or less sensitivity to detect insertions and deletions (in/dels), large CNVs, and structural chromosomal rearrangements (e.g., translocations, inversions). Local realignment after employing a global alignment strategy can help to more accurately call in/del variants.9 Large deletions and duplications are detected either by comparing actual read depth of a region to the expected read depth or through paired-end read mapping (independent reads that are associated to the same library fragment). Paired-end and mate-pair (joined fragments brought from long genomic distances) mapping can also be used to identify translocations and other structural rearrangements.
D.3.4. File formats. Many different formats exist for the export of raw variants and their annotations. Any variant file format must include a definition of the file structure and the organization of the data, specification of the coordinate system being used (e.g., the reference genome to which the coordinates correspond, whether numbering is 0-based or 1-based, and the method of numbering coordinates for different classes of variants), and the ability to interconvert to other variant formats and software. If sequence read data are provided as the product of an NGS test, they should conform to one of the widely used formats (e.g., “.bam” files for alignments, “.fastq” files for sequence reads) or have the ability to be readily converted to a standard format. Although there is currently no official gold standard, a de facto standard format that has emerged is the variant call format (.vcf) used by the 1000 Genomes Project. This structured-text file format conveys meta-information about a given file and specific data about arbitrary positions in the genome. It should be noted that the .vcf file format is typically limited to variant calls, and many advocate the inclusion of reference calls in the .vcf format to distinguish the absence of data from wild-type sequence. At the time of this publication, an effort led by the Centers for Disease Control and Prevention is currently under way to develop a consensus gVCF file format.
D.4. Variant filtering
In traditional disease-targeted testing, the number of identified variants is small enough to allow for the individual assessment of all variants in each patient, once common benign variations are curated. However, ES identifies tens of thousands of variants, whereas GS identifies several million, making this approach to variant assessment impossible. A filtering approach must be applied for ES/GS studies, and laboratories may even need to employ auto-classification strategies for very large disease-targeted panels. Regardless of the approach, laboratories should describe their methods of variant filtering and assessment, pointing out their limitations.
D.4.1. Disease-targeted panels. As Mendelian disease-targeted panels increase in gene content, laboratories may need to develop auto-classification tools to filter out common benign variants from rare highly penetrant variants. Sources of broad population frequencies that can be used for auto-classification of benign variation include dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP), the NHLBI Exome Sequencing Project (evs.gs.washington.edu/EVS), and the 1000 Genomes Project (http://www.1000genomes.org). The frequency cutoff that a laboratory employs for this step will depend on the maximum frequency of a single disease variant. The frequency is based on disease prevalence, inheritance pattern, and mutation heterogeneity. However, frequency cutoffs should be higher than the theoretical maximum to account for statistical variance in those population estimates due to the control sample size; the possibility of undocumented reduced penetrance; and the possible inclusion of individuals who have not been phenotyped, who have asymptomatic or undiagnosed disease, or who have a known disease. Furthermore, laboratories should not simply use currently available disease-specific databases for directly filtering variants to determine which will be reported as disease causing. Few, if any, variant databases are curated to a clinical grade with strict, evidence-based consensus assessment of supporting data. It is well known that many databases contain misclassified variants, particularly benign variation misclassified as disease causing.10 In addition, most Mendelian diseases have a large percentage of variants that are private (unique to families), requiring a robust process for assessing novel variation. Published guidelines contain further recommendations on the classification of sequence variants.11
D.4.2. Exome and genome sequencing. In the analysis of ES/GS, the assumptions that causative mutations for Mendelian disorders will be rare and highly penetrant must be made.1 However, the laboratory must apply additional strategies to variant and gene filtration beyond this assumption. Successfully identifying the molecular basis for a rare disorder may depend on the strategy employed, such as choosing appropriate family members for comparison, given a suspected mode of inheritance. The strategies employed will depend on the indication for testing and the intent to return incidental findings. The ultimate goal is to reduce the number of variants needing examination by a skilled analyst. Variants may be included or excluded based on factors including: presence in a disease candidate gene list, presumed inheritance pattern in the family (e.g., biallelic if recessive), likelihood of consanguinity in the parents (e.g., homozygous variants), mutation types (e.g., truncating, copy number), presence or absence in control populations, observation of de novo occurrence (if the phenotype is sporadic in the context of a dominant disorder), gene expression pattern, algorithmic scores for in silico assessment of protein function or splicing impact, and biological pathway analysis. The choice of filtering algorithm design may differ across case types and requires a high level of expertise in genetics and molecular biology. This expertise should include a full understanding of the limitations of the databases against which the patients results are being filtered and the limitations of both the sequencing platform and multiple software applications being used to generate the variants being evaluated. Individuals leading these analyses should have extensive experience in the evaluation of sequence variation and evidence for disease causation, as well as an understanding of the molecular and bioinformatics pitfalls that could be encountered.
Laboratories must balance overfiltering, which could inadvertently exclude causative variants, with underfiltering, which presents too many variants for expert analysis. A stepwise approach may be necessary, using a first pass to identify any clear and obvious causes of disease. If needed, filtering criteria can be subsequently reset to provide an expanded search, resulting in a larger number of variants for evaluation. Once the data analysis approach for each patient has been set, laboratories must provide ordering health-care providers documentation of their general and patient-specific processes. There should be clear documentation of the basic steps taken to achieve the reported results on each patient.
D.5. Sequencing regions with homology
Homologous sequences such as pseudogenes pose a challenge for all short-read sequencing approaches. Cocapture cannot be avoided for targeted NGS when a hybridization-based enrichment method is used. The limited length of NGS sequence reads can lead to FP variant calls when reads are incorrectly aligned to a homologous region, but also to false-negative (FN) results when variant-containing reads align to homologous loci. Therefore, the laboratory must develop a strategy for detecting disease-causing variants within regions with known homology. These strategies might include local realignment after employing a global alignment strategy, which can help with some types of misalignment due to homologous or repetitive sequences. In addition, paired-end sequencing may partially remedy this problem because reads can usually be mapped uniquely if one of the paired ends maps uniquely. Alternatively, if there is enough depth of coverage and relaxed allelic fraction ratios allow reads to be mapped to both homologous regions, then correct variant mapping can be elucidated with Sanger confirmation using uniquely complementary primer pairs.
D.6. Companion technologies and result confirmation
Result confirmation is essential when the analytic FP rate is high or not yet well established, particularly as in ES and GS approaches. Confirmation can also be used to confirm sample identity, which is critical when laboratory workflows are complex and not fully automated. FP rates for most NGS platforms in current use are appreciable, and therefore it is recommended that all disease-focused and/or diagnostic testing include confirmation of the final result using a companion technology. This recommendation may evolve over time as technologies and algorithms improve. However, laboratories must have developed extensive experience with NGS technology and be sufficiently aware of the pitfalls of the technologies they are using and the analytical pipelines developed before deciding that result confirmation with orthogonal technology can be eliminated, particularly for in/del variants, which are notably more challenging to detect and define correctly. Extensive validation of variant detection using all types of variation and across all variations in assay performance is necessary before confirmation can be eliminated or reduced. Sanger sequencing is most often employed as the orthogonal technology for germline nuclear DNA testing. For testing involving low-percentage variants such as the detection of somatic variants from tumor tissue, heteroplasmic mitochondrial variants, and germline mosaic variants, other approaches such as replicate testing may be necessary.
Sanger sequencing can also be used to “fill in” missing data from bases or regions that are supported by an insufficient number of reads to call variants confidently. It may be acceptable to report results without complete coverage at a predefined minimum for some tests, such as ES/GS and certain broad gene-based panels (e.g., large-scale recessive-disease carrier testing). The laboratory director has discretion to judge the need for Sanger sequencing to fill in missing areas of a test. However, it is currently recommended that all disease-focused testing of high-yield genes include complete coverage in each patient tested. It is also important, particularly for targeted gene panels, that the laboratory’s test report include information about regions/genes that were not covered in a given sample.
It is recommended that the companion assays required for this step be developed or planned for in advance of their need, to ensure reasonable turnaround times. Projected turnaround times should take into account the time required for using these companion technologies. In testing environments where confirmation of all results may not be possible before initial reporting (e.g., certain lower-risk incidental findings from ES/GS studies such as pharmacogenetic alleles for drugs not under consideration for the patient), it is recommended that laboratories clearly state the need for follow-up confirmatory testing. For example, the report could state “The following results have not been confirmed by an alternative method or replicate test, and the reported accuracy of variants included in this report is ~x% (or ranges from x to y%). If these results will be used in the care of a patient, the need for requesting confirmation must be weighed against the risk of an erroneous result.”
E. Test Development and Validation
E.1. General considerations
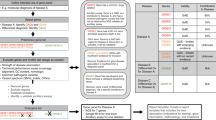
Various combinations of instruments, reagents, and analytical pipelines may be used in tests involving NGS. Some may in the future be approved by the US Food and Drug Administration (FDA) and be available through a commercial source. Other components may be labeled “for investigational use only,” “for research use only,” or as an “analyte-specific reagent.” In some instances, components are commercially available but must then be validated by the clinical laboratory for use as a diagnostic tool. Alternatively, components can be combined by the laboratory into a test and then validated within the laboratory for use as a diagnostic tool. Depending on the intended clinical application of the products, each may be subject to different levels of validation. Currently, any laboratory test that is not exclusively based on a FDA–approved assay is considered to be part of a laboratory developed test and will require a full validation (instead of a verification).12 The availability of validation data from outside sources may influence the extent to which a laboratory independently validates the products. However, the laboratory director must conduct an appropriate validation of each test offered in the clinical setting. The entire test development and validation process is shown in Figure 2 .

Next-generation sequencing test development and validation process. CNV, copy-number variant; in/dels, insertions and deletions; sample prep, sample preparation.
E.1.1. Test development and platform optimization. Once the individual components of the testing process (library preparation method, target capture if applicable, sequencer, and analysis tools) have been chosen, iterative cycles of performance optimization typically follow until all assay conditions as well as analysis settings are optimized. During this phase, if pooling of samples is planned, the laboratory should also determine the number of samples that can be pooled per sequencing run to achieve the desired coverage level and establish baseline cost and turnaround time projections. Due to the complexity of the data analysis process and the challenges surrounding correct mapping of short sequence reads, the laboratory should establish performance of the variant calling pipeline by analyzing data containing known sequence variants of various types (e.g., single-nucleotide variants, small in/dels, large CNVs, structural variants). The use of synthetic variants can help to create a rich set of testing data that can be used to compare various tools and to optimize settings and thresholds. A protocol for the entire workflow must be established and adhered to before proceeding to test validation. Optimization must include all sample types that will be evaluated in clinical practice (e.g., whole blood, saliva, formalin-fixed paraffin-embedded tissue).
E.1.2. Test validation. Once assay conditions and pipeline configurations have been established, the entire test should be validated in an end-to-end manner on all permissible sample types. Assay performance characteristics including analytical sensitivity and specificity (positive and negative percent agreement of results when compared with a gold standard) as well as the assay’s repeatability (ability to return identical results when multiple samples are run under identical conditions) and reproducibility (ability to return identical results under changed conditions) need to be established.12,13,14,15 Because NGS technologies are still relatively new and multiple options exist for every step in the workflow, the scope of validation depends on the degree to which analytic performance metrics have already been established for the chosen combination of sample preparation, sequencing platform, and data analysis method. The first test developed by a laboratory may therefore carry a higher “validation burden” than subsequent tests developed on an established platform using the same basic pipeline design. In practice, this may entail sequencing a larger number of samples to cover sufficient numbers of all variant types. In subsequent test validations, fewer samples of each type may be required. To determine the analytic validity of a test, the laboratory should utilize well-characterized reference samples for which reliable Sanger sequencing data exist. These samples should ideally be a renewable resource, which can then be used to establish baseline data with which future test modifications can be compared. See section F.7 for a more detailed discussion. Reference samples do not need to contain specific pathogenic variants because they are used to assess the overall ability of a test to detect a type of variant, and the clinical significance of the variant has little bearing on its detectability (analytic validity).
In addition to the general approaches described above, there are also content- and application-specific issues that must be addressed as noted below for targeted panels and exome and genome testing. For disease-specific targeted gene panels, test validations must include unique gene- and disease-specific aspects. It is critical to include common pathogenic variants in the validation set to ensure that the most common causes of disease are detectable, given that sequence-specific context can affect the detection of a variant. In addition, issues related to accurate sequencing of highly homologous regions need to be addressed when one or more genes within the test have known pseudogenes or other homologous loci.
For ES and GS, the focus of validation is shifted more toward developing metrics that define a high-quality exome/genome such as the average coverage across the exome/genome and the percentage of bases that meet a set minimum coverage threshold. Validation of ES/GS should include evaluation of a sample that was well analyzed using another platform. There are several samples available for this purpose that have previously been sequenced using Sanger methods. See section F.7 for a more detailed discussion. For GS, the sample could be one that was previously analyzed using a high-density single-nucleotide polymorphism (SNP) array. However, this approach is less useful for ES because most of the SNPs on commercially available high-density SNP arrays are not in regions targeted by ES capture kits. An evaluation of the concordance of SNPs identified as compared with the reference should be made for either ES or GS (some laboratories have used 95–98% concordance as the minimum acceptable level).
In addition, validation of ES and GS should include sequencing a variety of samples containing previously identified variants. This part of the validation process is identical to what is recommended for targeted tests and should establish analytical performance for a wide variety of variants and variant types to ensure maximum confidence in the ability of the test to identify rare and novel variants. The samples should be blinded and subjected to the entire end-to-end test protocol, including the bioinformatics pipeline that will be used for the ES/GS test. Samples should be selected that represent the full spectrum of mutation types to be analyzed.
E.1.3. Platform validation. Performance data across all tests developed by a laboratory with the same basic platform design can be combined to establish a cumulative “platform” performance. By maximizing the number and types of variants tested across a broad range of genomic regions, confidence intervals can be tightened. Because the size of NGS tests make validation of every base impossible, this approach enables extrapolation of performance parameters to novel-variant discovery within the boundaries of the established confidence.
E.1.4. Quality management. Finally, the laboratory must develop quality control (Q/C) measures and apply these to every run. These can vary depending on the chosen methods and sequencing instrument but typically include measures to identify sample-preparation failures as well as measures to identify failed sequencing runs. The laboratory must also track sample identity throughout the testing process, which is especially important given that NGS testing commonly entails pooling of barcoded samples. Proficiency testing (PT) protocols must also be established and executed periodically according to Clinical Laboratory Improvement Amendments (CLIA) regulations.
E.2. Data analysis optimization
Analysis of data generated on NGS platforms is complex and typically requires a multistage data handling and processing pipeline. Given the use of separate tools for data analysis, which are independent of the wet-laboratory steps, and the high likelihood of laboratory customizations of these data analysis tools, it is recommended that the analytical pipelines be validated separately during initial test development. Later, the prevalidated pipeline can then be included in each end-to-end test validation, which includes both the wet-laboratory steps and the analytical pipeline. If using commercially developed software, the laboratory should make all attempts to document any validation data provided by the vendor, but the laboratory must also perform an independent validation of the tool.
The laboratory must determine the parameters and thresholds necessary to determine whether the overall sequencing run is of sufficient quality to be considered successful. This may include analyses at intermediate points during the sequencing run as well as at the completion of the run (e.g., real-time error rate, percentage of target captured, percentage of reads aligned, fraction of duplicate reads, average coverage depth, range of insert size). In addition, the laboratory should set and track thresholds for coverage to ensure sufficient coverage is achieved for variant calling, as well as allelic fraction, which influences analytical sensitivity and specificity. Note that NGS variant calling tools apply default thresholds, which may have to be optimized to enhance analytic performance. In addition, the laboratory must establish that the analytical pipeline can accurately track sample identity, particularly if barcoding is used.
E.2.1. Coverage. Generally, variant calls are more reliable as coverage increases, and for ES/GS when trios are sequenced. Low coverage increases the risks of missing variants (FNs) and assigning incorrect allelic states (zygosity), especially in the presence of amplification bias, and decreases the ability to effectively filter out sequencing artifacts, leading to FPs. Laboratories should establish a minimum coverage threshold necessary to detect variants based on their diagnostic approach (e.g., only proband sequenced, proband plus parents sequenced for ES/GS) and report analytical performance related to the minimum threshold that is guaranteed for the test. For the detection of germline heterozygous variants, some laboratories use 10–20X as a minimum for covering all bases of a targeted panel. For ES and GS testing, it may be more useful to track minimum mean coverage as well as the percentage of bases that reach an absolute minimum threshold. For example, a laboratory might ensure that ES reaches a minimum mean coverage of 100X for the proband and 90–95% of bases in the laboratory’s defined target reach at least 10X coverage. A lower threshold of 70X might be used when trios are sequenced. For GS, a laboratory might ensure that the assay reaches a minimum mean coverage of 30X. Higher coverage, as well as additional variant calling parameters, is required for the detection of variants from mixed or mosaic specimens (e.g., somatic tumor samples with a low percentage of tumor cells, mitochondrial heteroplasmy, germline mosaicism). It is also important to note that minimum coverage is highly dependent on many aspects of the platform and assay including base-call error rates, quality parameters such as how many reads are independent versus duplicate, and other factors such as analytical pipeline performance. Therefore, it is not possible to recommend a specific minimum threshold for coverage, and laboratories will need to choose minimum coverage thresholds in accordance with total metrics for analytical validation.
E.2.2. Allelic fraction and zygosity. Germline heterozygous variants are expected to be present in 50% of the reads. However, amplification bias and low coverage can lead to a wider range. Laboratories must determine allelic fraction ranges to (i) distinguish true calls from FP calls, which typically have a low allelic fraction, and (ii) assign zygosity. The chosen parameters may be influenced by the inclusion of a confirmation step. Parameters that give higher sensitivity but lower specificity may be chosen when Sanger confirmation testing will be performed (e.g., for targeted testing and primary findings of ES/GS). However, thresholds with higher specificity are recommended for calling variants that may be included in incidental findings without confirmation. It is recommended to analyze the performance of different types of variants separately because their performance may vary. For example, coverage and allelic fraction for in/dels can be lower when the alignment tool discards in/del-containing reads.
Additional metrics that may be helpful for determining data quality include the percentage of reads aligned to the human genome, the percentage of reads that are unique (before removal of duplicates), the percentage of bases corresponding to targeted sequences, the uniformity of coverage, and the percentage of targeted bases with no coverage.
E.3. Determination of performance parameters
The validation process should document: (i) analytical sensitivity and FN rate, (ii) analytical specificity and FP rate, (iii) predicted clinical sensitivity, and (iv) assay robustness and reproducibility. Parameters may be calculated at the technology level initially; however, if multiple technologies are used in the test, the final reported parameters should be relevant to the full protocol of the test. For example, tests may include both NGS and companion technologies including steps to confirm variants and/or fill in missing data. The final analytical parameters of a test must reflect the entire testing process.
E.3.1. Analytical sensitivity. Current American College of Medical Genetics and Genomics (ACMG) guidelines often define analytical sensitivity as the “proportion of biological samples that have a positive test result or known mutation and that are correctly classified as positive” but also state that this concept does not fit tests that use genome scanning methods where novel, unclassified variants can be detected (http://www.acmg.net; Standards and Guidelines for Clinical Genetic Laboratories 2008 Edition, section C8.4). These tests therefore require a dual approach. For a given test, the laboratory must document that the NGS assay can correctly identify known disease-causing variants, particularly if they are common in the tested population. To project the analytical sensitivity for detecting novel variants, it is necessary to extrapolate from the analysis of known variants to the entire region analyzed. Here, it is important to maximize the number of variants tested as well as the genomic regions they represent and calculate confidence intervals. An online tool for calculating confidence intervals can be found at http://www.pedro.org.au/english/downloads/confidence-interval-calculator/. It is recommended that a separate calculation be performed for each variant type that is relevant for the clinical context in which testing is offered (e.g., substitutions, in/dels, CNVs). It is well known that detectability of variants can be influenced by local sequence context, and therefore a high general sensitivity may not always be true for every possible variant. However, the higher the number of variants tested and the larger and more diverse the genomic loci included in this cumulative analysis, the higher the confidence that the established sensitivity can be accurately extrapolated.14 The variants included in this type of analysis do not have to be pathogenic because this has no bearing on their detectability.
E.3.2. Analytical specificity. Current ACMG guidelines define analytical specificity as “the proportion of biological samples that have a negative test result or no identified mutation (being tested for) and that are correctly classified as negative” (http://www.acmg.net; Standards and Guidelines for Clinical Genetic Laboratories 2008 Edition, section C8.4). Traditionally, “negative” is defined as the absence of a pathogenic variant; however, for the reasons described above, this is not a meaningful measure to define analytical performance in sequencing tests. By contrast, it is most useful to calculate the average FP rate per sample and then express this as number of FPs/interval tested (e.g., per kb of sequence). This will inform an estimate of the number of FPs expected per sample for a given test and will also allow an extrapolation for larger panels including the exome and genome. If variant calls are confirmed by Sanger sequencing, the technology-specific FP rate is less critical unless it generates an amount of confirmatory testing per sample that is not sustainable for the laboratory.
E.3.3. FN and FP rates. The FN rate can be calculated as 1 − sensitivity. The FP rate can be calculated as 1 − specificity.
E.3.4. Clinical sensitivity. For disease-specific targeted panels, the laboratory should establish the estimated clinical sensitivity of the test on the basis of a combination of analytical performance parameters and the known contribution of the targeted set of genes and types of variants detectable for that disease. For ES and GS of patients with undiagnosed disorders, it is not feasible to calculate a theoretical clinical sensitivity for the test given its dependency on the applications and indications for testing. However, empirical data from one recent study suggest that these tests have a clinical sensitivity of ~20%.3 Likewise, laboratories should track and share success rates across different disease areas to aid in setting realistic expectations for the likelihood of an etiology being detected for certain types of indications.
E.3.5. Assay robustness. It is recommended that laboratories measure robustness (likelihood of assay success) for the main assay components such as library preparation and sequencing runs and have adequate Q/C measures in place to assess their success (see section F.4).
E.3.6. Assay precision. It is recommended that the laboratory document the assay’s precision (repeatability and reproducibility) based on known sources of variation. For example, it is suggested that the laboratory take a single library or sample preparation and run it on two or three lanes/wells within the same run (repeatability, within-run variability) as well as on two or three different runs (reproducibility, between-run variability). Other meaningful measures of reproducibility are instrument-to-instrument variability (run samples on two or three different instruments if available) and interoperator variability. Complete concordance of results is unlikely for NGS technologies; however, the laboratory should establish parameters for sufficient repeatability and reproducibility. For example, a range of >95–98% has been used by some laboratories.
E.3.7. Limit of detection. It is recommended that laboratories determine the minimal specimen requirements to generate enough DNA to complete the assay and any follow-up analysis. In addition, if testing samples with mixed content (tumor, mitochondrial, mosaic, etc.), the laboratory must determine the lower limit of detection of variants based on dilution assays, mixing two pure samples at variable percentages. This value should be used in providing a lower limit of detection for likely mixed specimens as well as acceptance criteria for tumor specimens with assessed tumor percentage.
E.4. Validating modified components of a test or platform
E.4.1. Modified assay conditions, reagents, instruments, and analytical pipelines. Because NGS is a dynamic technology, suppliers are continually improving the chemistries. Laboratories must validate any changes to the existing test (e.g., new sequencing chemistry, new instrument, new lot of capture reagents, new software versions) using an end-to-end test validation with previously analyzed specimens or well-characterized controls. It is recommended to always include the same, renewable, well-characterized sample (e.g., a HapMap sample) and determine analytical performance parameters and other parameters, such as coverage, as outlined in the prior sections.
E.4.2. Added/modified test content. When adding new genes to an established targeted panel, the laboratory should analyze a small number of samples and establish that the performance of the test for analyzing the existing genes has not been altered and that the performance for analyzing the new genes is acceptable.
The above-described changes should be reviewed by the laboratory director. Separate documentation should be generated, and the date of introduction of the new version into the pipeline for clinical samples should be documented.
F. Analytical Standards
Monitoring preanalytical variables, analytical variables, and postanalytical variables should be part of the laboratory’s quality assurance (Q/A) and quality improvement programs. Such variables may include quality of the specimen received, number of NGS run failures, and variant detection parameters.
F.1. Specimen requirements
NGS may be performed on any specimen that yields DNA (e.g., peripheral blood, fresh or frozen tissues, paraffin-embedded tissues, prenatal specimens). The laboratory needs to establish the types of specimens (which may include acceptance of genomic DNA as a sample type) and minimum required quantities appropriate for NGS assays. The quality of DNA and variant detection requirements will likely differ for some specimen types, and, as such, the laboratory will need to determine acceptable parameters for each sample type (e.g., volume, amount of tissue).
F.2. DNA requirements and processing
The laboratory should establish the minimum DNA requirements to perform the test. Considerations include whether the test is performed once per specimen and how much DNA may be required for confirmatory and follow-up procedures. The laboratory should have written protocols in the laboratory procedure manual and/or quality management program for DNA extraction and quantification (e.g., fluorometry, spectrophotometry) for obtaining an adequate quality and concentration of DNA. The laboratory should have documentation of these parameters in each patient record.
F.3. Suboptimal samples
If a sample does not meet requirements of the laboratory and is deemed suboptimal, the recommended action is to reject the specimen and request a new specimen. If obtaining a new specimen is not possible, whole-genome amplification could be considered if the laboratory is experienced in this technique. In this case, the potential biases inherent in the technique (e.g., uneven or incomplete amplification of the entire genome) should be detailed in the report so that the physician and patient are informed of the limitations of this technique. Written standards describing when and how the whole-genome amplification procedure is performed should be incorporated into the laboratory manual.
F.4. Quality control and quality assurance
A Q/A program used by the laboratory is expected to support the routine analysis of samples and the interpretation and reporting of NGS data. Laboratories should put in place predetermined Q/C checkpoints for monitoring Q/A. The Q/A program should also include documentation of which instruments are used in each test and documentation of all reagent lot numbers. Laboratories are expected to document any deviation from the standard procedures established by the laboratory during the validation process.
F.4.1. General quality control. The development of Q/C stops during the workflow is essential for any clinical laboratory test and of critical importance for many NGS platforms, because assays can be lengthy and expensive and identification of samples that have a high probability of generating results of unacceptable quality is important to ensure optimal turnaround times. However, due to the high diversity of NGS platforms and assays, specific Q/C measures may vary. Similar to Sanger-based sequencing, positive controls do not need to be tested concurrent with routine clinical tests;16 however, the operating procedure must have methods to evaluate and control for possible contamination at various points in the procedure. Generally, Q/C stops need to be added to the wet-laboratory process before the sequencing run, to the sequencing run itself, and at the end of the sequencing run before executing data analysis. Examples of Q/C stops include determining the success of initial DNA fragmentation (incompletely sheared gDNA will result in suboptimal data), monitoring error rates during the sequencing run (enabling abortion of the run if there is a problem), and postrun/preanalysis assessment of the read quality (e.g., percentage of bases above a predetermined quality threshold, Q/C alignments).
F.4.2. Bioinformatics. A Q/A program for the bioinformatics process or pipeline should be developed to support the analysis, interpretation, and reporting of NGS data. The Q/A program should also document corrective measures that have been put in place by the laboratory to report and resolve any deviation from the developed pipeline during the testing process.
The laboratory must also document the bioinformatics pipeline that it uses in the analysis of NGS data and capture the specific version of each component of the pipeline utilized in the analysis of each patient test. A system must be developed that allows the laboratory to track software versions, the specific changes each version incorporates, and the date the new version was implemented on clinical samples. The Q/A program should be developed to include the description of input and output files for each step of the process and metrics and Q/C parameters for optimal performance.
F.5. Staff qualifications
Given the technical and interpretive complexity of NGS, we recommend that the reporting and oversight of clinical NGS-based testing be performed by individuals with appropriate professional training and certification (American Board of Medical Genetics–certified medical/laboratory geneticists or American Board of Pathology–certified molecular genetic pathologists) and with extensive experience in the evaluation of sequence variation and evidence for disease causation as well as technical expertise in sequencing technologies. For laboratories offering ES or GS services, the laboratory should have access to broad clinical genetics expertise for evaluating the relationships between genes, variation, and disease phenotypes.
F.6. Data storage and traceability of patient reports
NGS generates a massive amount of data, and laboratories may choose to store the data in-house or offsite. Cloud computing is also becoming a widely available choice for data analysis and storage. However, many cloud computing environments are not Health Insurance Portability and Accountability Act compliant, and therefore laboratories must ensure that the method used for data storage is compliant with the Act and allows traceability of patient data. Due to the multistep nature of NGS informatics analysis, files with differing information contents and sizes will be generated. Generally, NGS sequencing image files, which can be several terabytes in size, are not stored. Laboratories may employ widely heterogeneous sequence alignment and variant calling algorithms; thus, the types of files generated in the process of NGS will differ greatly between laboratories. Laboratories should make explicit in their policies which file types and what length of time each type will be retained, and the data retention policy must be in accordance with local, state, and federal requirements. CLIA regulations (section 493.1105) require storage of analytic systems records and test reports for at least 2 years. For more specific suggestions for NGS technologies, we recommend that the laboratory consider a minimum of 2-year storage of a file type that would allow regeneration of the primary results as well as reanalysis with improved analytic pipelines (e.g., bam or fastq files with all reads retained). In addition, laboratories should consider retention of the VCF, along with the final clinical test report interpreting the subset of clinically relevant variants, for as long as possible, given the likelihood of a future request for reinterpretation of variant significance.
F.7. Reference materials
Reference materials (RMs) are used by clinical laboratories for test validation, Q/C, and proficiency testing (PT). The goal for NGS applications is to have genome-wide-characterized RMs for sequence and CNVs. This is important because no single clinical-grade reference sequence exists today. However, several human cell lines available from Coriell Repositories (e.g., NS12911; Levy et al.17) have had GS performed with published results.
The Centers for Disease Control and Prevention (http://www.cdc.gov/clia/Resources/GetRM) and the National Institute for Standards and Technology have efforts to more fully characterize several of the Coriell samples by organizing laboratories performing gene panel, ES, or GS, and copy-number variation analysis. Depth of coverage and consensus between laboratories and platforms will be recorded, so laboratories will know which areas of the genome are highly characterized and which do not yet have consensus. However, if cell lines are used as RM, genomic stability over time will be a concern. Studies including sequencing different passages will be necessary to understand the extent to which instability will affect the use of cell lines for NGS. In addition, several other organizations such as the College of American Pathologists, and the US Food and Drug Administration also have efforts to define RMs for use in evaluating instrumentation and test performance.
Genomic DNA extracted from blood is stable, but gathering enough from one individual for long-term, potentially multi-laboratory use is challenging. Still, it could be possible to have large quantities of several samples that could be used for years via whole-genome amplification.
Simulated electronic sequences created through computational methods may also be useful and could be incorporated into Q/C or PT processes. Simulated sequences are typically designed to focus on a specific region to address a specific issue, such as repetitive sequences, known in/dels, and SNPs.
F.8. Proficiency testing
CLIA requirements for PT or alternative assessments pose challenges for NGS. Typically, PT/alternative assessments are performed twice yearly for each clinically offered assay. For NGS, the definition of the assay may be a gene panel, exome analysis, or genome analysis. A formal proficiency challenge available for single-gene disorders from the Biochemical and Molecular Resource Committee (a joint committee of the College of American Pathologists and the ACMG) typically has two PT challenges yearly, each consisting of three samples. The current cost of performing a gene panel, ES, or GS assay on six PT samples per year may not be financially feasible for most clinical laboratories. Therefore, other models are being explored, such as methods-based (technical wet laboratory) or analytical (informatics) challenges. The College of American Pathologists/ACMG Sequencing survey currently involves a Sanger-sequencing challenge in which electronic files of sequences and references are sent to participants to assess their ability to align, detect, properly name, and interpret sequence variants in any gene. A similar challenge could be used for the informatics portion of an NGS assay. The College of American Pathologists released a laboratory checklist for NGS in July 2012 and is currently implementing a pilot PT program for NGS which is expected to be available widely in 2014. The European Quality Network has launched a pilot PT program that is also available to US laboratories. However, until PT services for NGS/ES/GS are fully available laboratories are required to employ existing acceptable PT approaches according to CLIA guidelines using national programs if available, interlaboratory exchange if no national program is available, and intralaboratory PT if no other laboratory performs an equivalent NGS test.
G. Reporting Standards
G.1. Turnaround times
The laboratory should have written standards for NGS test prioritization and turnaround times that are based on the indication for testing. These turnaround times should be clinically appropriate.
G.2. Data interpretation
Each variant identified in a disease-targeted test should be evaluated and classified according to ACMG guidelines.11 This should include an evidence-based assessment of the likelihood that the variant disrupts the function of the gene or gene product as well as its potential role in disease. For tests that cover a broad range of phenotypes, as well as for reporting results from ES and GS tests, in addition to evaluating whether the variant likely alters gene or protein function, the assessment should also evaluate whether the known phenotypes associated with gene disruption match the patient’s phenotype. If multiple variants of potential clinical significance are identified, the interpretation should discuss the likely relevance of each variant to the patient’s phenotype and prioritize variants accordingly.
For ES/GS reporting, it is at the discretion of the laboratory to decide whether to report variants in genes without any known disease association. If the laboratory accepts patients who have tested negative for existing disease-targeted tests, the laboratory should have a plan in place for how it will evaluate variants in genes without a known disease association. Patients should not be expected to bear the costs of research-grade analyses, and any results that are gained from these analyses should be presented as preliminary findings until evidence can be garnered that definitively supports the association of that gene with a human clinical phenotype.
Laboratories are also strongly encouraged to deposit data from clinical sequencing into public databases such as ClinVar (http://www.ncbi.nlm.nih.gov/clinvar), in order to more rapidly build knowledge that will lead to improved care.
G.3. Reporting of incidental findings
As an inevitable consequence of ES/GS testing, sequence information will be generated that is not immediately germane to the diagnostic intent of the test. Such incidental findings may or may not have clinical implications for a given patient, and some patients may not desire the return of such information. Variants of uncertain significance (VUSs) that are discovered as incidental findings in genes unrelated to the diagnostic evaluation should not be returned because they have no clear clinical implications and are more likely to cause confusion or harm. However, certain types of incidental findings may be deemed sufficiently “medically actionable” such that their return would be strongly encouraged. Therefore, it is recommended that the laboratory carefully develop a policy and process for returning such information and ensure that it conforms to accepted medical and ethical obligations as well as additional practice policies that will continually develop in this area. The laboratory should provide the following information about incidental findings: (i) whether it will systematically search for, and report on, certain findings, as set forth in the recent ACMG Recommendations for reporting of Incidental Findings in Clinical Exome and Genome Sequencing18 or whether it will only report variants that are uncovered unintentionally; (ii) whether incidental findings are routinely confirmed to ensure analytic accuracy or whether confirmation is recommended through additional follow-up testing (see section D.6); (iii) a clear definition of the criteria used to decide what types of incidental findings to report; and (iv) clear instructions on how all, or certain types of, incidental findings can be requested and whether and how they can be declined.
In this way, the ordering provider will be aware of the potential scope of incidental findings before ordering ES/GS testing and can ensure that informed consent and shared decision making with the patient includes a discussion of how incidental findings will be handled. Any return of incidental findings should be done in collaboration with the ordering provider to ensure that those results are interpreted in the context of the patient’s medical and family history and personal desires for receiving incidental results. Additional details on the return of incidental findings are outside the scope of this document but are available in a separate set of ACMG Recommendations.18
G.4. Written report
All NGS reports must include a list of variants identified, annotated according to HGVS nomenclature (http://www.hgvs.org) and clinically classified according to ACMG guidelines.11 These guidelines are updated regularly, and the laboratory should review these guidelines carefully. Gene names should adhere to the approved HUGO Gene Nomenclature Committee nomenclature (http://www.genenames.org). An example data structure is shown: MYBPC3 (NM_000256.3), heterozygous c.1504C>T (p.Arg502Trp), exon 17, pathogenic.
The transcript being used for providing c. and p. nomenclature and exon numbering should be provided in the report, either with the variant as noted above, in the methodology, or through a referenced Internet-accessible website. If a variant has a different nomenclature across different transcripts relevant to the indication for testing, the variant should be reported according to the major transcript unless a different, and potentially greater, impact is predicted for another transcript. In the latter case, both impacts should be described in the report.
It is recommended that for reporting the primary findings in a targeted diagnostic test, a succinct, high-level interpretive result should be provided at the start of the interpretation consisting of findings that are “positive” (detection of a mutation that explains a patient’s condition), “negative” (no variants identified of likely relevance to the diagnostic indication), and “inconclusive” (a clear explanation of the patient’s condition was not found either due to only variants of unknown significance being identified or due to only a single heterozygous variant identified for a recessive condition). Other overall result interpretations may be appropriate for certain indications such as “carrier” for recessive carrier screening tests. Additional one-line explanations can be added as noted in the sample reports (see Supplementary Data online). It is also recommended that variants be listed according to their relevance to the patient’s indication for testing.
Laboratories should document the supporting evidence used to classify variants with respect to their known or potential role in disease. It is at the discretion of the laboratory to report variants classified as “likely benign” or “benign.” Laboratories must document a clear policy for determining what variants are excluded from reports in both material provided to ordering providers and on the patient’s individual report. For targeted NGS tests, if likely benign or benign variants are included, they should be clearly delineated from variants with known or more likely clinical relevance (e.g., pathogenic and VUSs). Laboratories may also wish to separate variants with known or assumed pathogenicity from VUSs. For genomic and exomic sequencing, VUSs should be reported if found in genes relevant to the primary indication for testing but should not be reported if found in genes outside those relevant to the primary indication for testing as described in the preceding section on the reporting of incidental findings.
It is recommended that laboratories report variant data in structured format, according to evolving health-care information technology standards. Current standards have been developed for HL7 messaging and dictate a structure for variant reporting (HL7 version 2 Implementation Guide: Clinical Genomics at http://www.hl7.org/implement/standards). The variant elements should include gene name, zygosity, cDNA nomenclature, protein nomenclature, exon number, and clinical assertion as noted above. This structure enables deposition of variants into the electronic health record, which in turn enables clinical decision–support algorithms to be leveraged for effective use of genetic information in health care. However, it is currently recommended that genetic data entering the electronic health record environment be restricted to those results relevant to the indication for testing and incidental findings with evidence of both analytical and clinical validity.
For targeted NGS tests, the report should contain a summary of the genes analyzed, and if full coverage is not guaranteed for all genes, then actual coverage achieved across the targeted region must be provided in each patient report. It should be noted that complete coverage of all high-yield genes, using Sanger sequencing to fill in missing regions, is strongly recommended. In addition, for negative test results from targeted analyses, the diagnostic yield (empiric or predicted) of the panel of genes analyzed should be provided to assist in determining the clinical sensitivity of the test. The laboratory should also report any limitations in analysis for specific variant types (such as CNVs) if the method of analysis does not include all variant types.
For ES/GS testing, a description of the process of data analysis should be provided in the report, whether the result is positive, negative, or inconclusive. Technical parameters regarding the level of coverage of the exome or genome should be provided. For GS, in addition to genome coverage, a separate coverage value for gene coding regions should be provided. In addition, if the laboratory is asked to perform analysis for a phenotype with one or more established causative genes, the gene coverage should be reported as well as any additional limitations related to the analytical detection of reported variants for the provided phenotype.
If parents or other family members are tested to assist with the interpretation of the variants found in the proband (e.g., submission of a trio of samples), only the minimal amount of information required to interpret the variants and comply with Health Insurance Portability and Accountability Act regulations should be provided in the proband’s report. Specific names and relationships should be avoided if possible. As an example, the following statements would be appropriate: “Parental studies demonstrate that the variants are on separate copies of the gene, with one inherited from each parent.” “Segregation studies showed consistent inheritance of the variant with the disease in three additional affected family members.”
Sample reports are included as examples of some ways to provide the content recommended above (see Supplementary Data online). Additional details in these sample reports are provided as examples only, and such details are ultimately left to the discretion of the laboratory director. However, reports must be clear and concise with the clinical significance of any relevant findings clearly stated and comprehensible for all varieties of health-care providers.
G.5. Data reanalysis
As the content of sequencing tests expands and the number of variants identified grows, expanding to thousands and millions of variants from ES and GS, the ability for laboratories to update reports as variant knowledge changes will be untenable without appropriate mechanisms and resources to sustain those updates. To set appropriate expectations with physicians and patients, laboratories should provide clear policies on the reanalysis of data from genetic testing and whether additional charges may apply for reanalysis.
For reports containing VUSs related to the primary indication and in the absence of updates that may be proactively provided by the laboratory, it is recommended that laboratories suggest periodic inquiry by physicians to determine if knowledge has changed on any VUSs including variants reported as “likely pathogenic.” Please see ACMG guidelines on duty to recontact regarding physician responsibility.19
H. Summary
Identifying disease etiologies for genetic conditions with substantial genetic heterogeneity has been a long-standing and challenging diagnostic hurdle. NGS overcomes many of the scalability obstacles for large-scale sequencing of DNA that have been faced by clinical laboratories utilizing traditional Sanger methods. However, along with the capability to produce high-quality sequence data for applications ranging from clinically relevant targeted panels to the whole genome, NGS brings new technical challenges that must be appreciated and logically addressed. This first version of the ACMG Clinical Laboratory Standards for Next-Generation Sequencing covers a broad spectrum of topics for those already offering diagnostic testing based on this technology as well as those considering their options for how to enter this arena. Most of the topics should be familiar to this audience but are discussed in some detail given the many unique circumstances and demands of NGS. Although key aspects of the clinical implementation of NGS technology have been addressed, additional recommendations regarding specific applications of the technology may be needed in the future. As always, the diagnostic community will collectively benefit by discussing the newest and most pressing NGS issues together. This will require an ongoing dialogue among those already engaged in this pursuit, those determining how to become involved in this new paradigm of molecular testing, and those who will be responsible for ordering and communicating NGS results to patients.
Disclosure
H.L.R., S.J.B., P.B.-T., K.K.B., J.L.D., M.J.F., B.H.F., M.R.H., and E.L. are employed by fee-for-service laboratories performing next-generation sequencing (NGS). Several individuals serve on advisory boards or in other capacities for companies providing NGS services (H.L.R.: BioBase, Clinical Future, Complete Genomics, GenomeQuest, Illumina, Ingenuity, Knome, Omicia; S.J.B.: GenomeQuest, EdgeBio; M.R.H.: GenomeQuest, RainDance; B.H.F.: InVitae). The other authors declare no conflict of interest.
References
Majewski J, Schwartzentruber J, Lalonde E, Montpetit A, Jabado N . What can exome sequencing do for you? J Med Genet 2011;48:580–589.
Cirulli ET, Singh A, Shianna KV, et al.; Center for HIV/AIDS Vaccine Immunology (CHAVI). Screening the human exome: a comparison of whole genome and whole transcriptome sequencing. Genome Biol 2010;11:R57.
Gahl WA, Markello TC, Toro C, et al. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med 2011;14:51–59.
Mamanova L, Coffey AJ, Scott CE, et al. Target-enrichment strategies for next-generation sequencing. Nat Methods 2010;7:111–118.
Glenn TC . Field guide to next-generation DNA sequencers. Mol Ecol Resour 2011;11:759–769.
Clark MJ, Chen R, Lam HY, et al. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol 2011;29:908–914.
Ledergerber C, Dessimoz C . Base-calling for next-generation sequencing platforms. Brief Bioinformatics 2011;12:489–497.
Li H, Homer N . A survey of sequence alignment algorithms for next-generation sequencing. Brief Bioinformatics 2010;11:473–483.
DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498.
Bell CJ, Dinwiddie DL, Miller NA, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med 2011;3(65):65ra64.
Richards CS, Bale S, Bellissimo DB, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med 2008;10(4):294–300.
Jennings L, Van Deerlin VM, Gulley ML ; College of American Pathologists Molecular Pathology Resource Committee. Recommended principles and practices for validating clinical molecular pathology tests. Arch Pathol Lab Med 2009;133:743–755.
Chen B, Gagnon M, Shahangian S, Anderson NL, Howerton DA, Boone JD . Good laboratory practices for molecular genetic testing for heritable diseases and conditions. MMWR Recomm Rep 2009;58(RR-6):1–37; quiz CE-31-34.
Mattocks CJ, Morris MA, Matthijs G, et al.; EuroGentest Validation Group. A standardized framework for the validation and verification of clinical molecular genetic tests. Eur J Hum Genet 2010;18:1276–1288.
Gargis AS, Kalman L, Berry MW, et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat Biotechnol 2012;30:1033–1036.
Maddalena A, Bale S, Das S, Grody W, Richards S ; ACMG Laboratory Quality Assurance Committee. Technical standards and guidelines: molecular genetic testing for ultra-rare disorders. Genet Med 2005;7:571–583.
Levy S, Sutton G, Ng PC, et al. The diploid genome sequence of an individual human. PLoS Biol 2007;5:e254.
Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013;15:565–574.
Hirschhorn K, Fleisher LD, Godmilow L, et al. Duty to re-contact. Genet Med 1999;1:171–172.
Acknowledgements
Members of the ACMG Working Group on Next-Generation Sequencing were all reviewed for conflicts of interest by the Board of the ACMG. This document was approved by the ACMG Board of Directors, 22 April 2013. H.L.R. was supported in part by National Institutes of Health grant HG006500.
Author information
Authors and Affiliations
Consortia
Corresponding author
Supplementary information
Supplementary Data
(PDF 45 kb)
Rights and permissions
About this article
Cite this article
Rehm, H., Bale, S., Bayrak-Toydemir, P. et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med 15, 733–747 (2013). https://doi.org/10.1038/gim.2013.92
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2013.92
Keywords
This article is cited by
-
Whole-genome sequencing of multiple related individuals with type 2 diabetes reveals an atypical likely pathogenic mutation in the PAX6 gene
European Journal of Human Genetics (2023)
-
Ethics in pre-ART genetics: a missed X-linked Menkes disease case
Journal of Assisted Reproduction and Genetics (2023)
-
X-CAP improves pathogenicity prediction of stopgain variants
Genome Medicine (2022)
-
Clinical pharmacogenetic analysis in 5,001 individuals with diagnostic Exome Sequencing data
npj Genomic Medicine (2022)
-
Performance Evaluation of Three DNA Sample Tracking Tools in a Whole Exome Sequencing Workflow
Molecular Diagnosis & Therapy (2022)
