
Abstract
Purpose:
Exome sequencing of a single individual for a clinical indication may result in the identification of incidental deleterious variants unrelated to the indication for testing (secondary findings). Given the recent availability of clinical exome testing, there is a limited knowledge regarding the disclosure preferences and impact of secondary findings in a clinical diagnostic setting. In this article, we provide preliminary data regarding the preferences for secondary findings results disclosure based on the first 200 families referred to Ambry Genetics for diagnostic exome sequencing.
Methods:
Secondary findings were categorized into four groups in the diagnostic exome sequencing consent: carrier status of recessive disorders, predisposition to later-onset disease, predisposition to increased cancer risk, and early-onset disease. In this study, we performed a retrospective analysis of patient responses regarding the preferences for secondary findings disclosure.
Results:
The majority of patients (187/200; 93.5%) chose to receive secondary results for one or more available categories. Adult probands were more likely than children to opt for blinding of secondary data (16 vs. 4%, respectively). Among responses for blinding, preferences were evenly scattered among categories.
Conclusion:
These data represent the unprecedented results of a large reference laboratory providing clinical exome sequencing. We report, for the first time, the preferences of patients and families for the receipt of secondary findings based on clinical genome sequencing. Overwhelmingly, families undergoing exome sequencing opt for the disclosure of secondary findings. The data may have implications regarding the development of guidelines for secondary findings reporting among patients with severe and/or life-threatening disease undergoing clinical genomic sequencing.
Genet Med 16 5, 395–399.
Similar content being viewed by others


Introduction
Next-generation sequencing technologies allow for sequencing of the exome and genome at a rapid rate, generating a large amount of genomic data. In 2012, the American College of Medical Genetics and Genomics (ACMG) published a statement regarding the clinical application of genomic sequencing stating that “The ACMG recognizes that genomic sequencing approaches can be of great value in the clinical evaluation of individuals with suspected germline genetic disorders… results will provide an explanation for a patient’s phenotype by identifying a mutation in a gene known to be associated with the patient’s clinical condition or in a gene that is highly likely to be causative given current knowledge”1 (Supplementary Table S1 online). For each exome sequenced, there are thousands of variants identified unrelated to the disease of interest. Although the vast majority of these alterations are likely benign polymorphisms or within clinically novel genes whose function remains largely obscure, a small number (<5) of well-described disease-association mutations are typically detected (unpublished data). For example, nonbiased massively parallel sequencing-based carrier-screening assays show that every individual carries an average of 2.8 mutations among genes known to cause severe pediatric disease in the recessive form.2 Among 37 well-described cancer predisposition genes, ~1% of individuals manifest a likely pathogenic cancer predisposition gene alteration.3 Although secondary to the referral indication for exome sequencing, these variants may also have important clinical and medical consequences for the patient including presymptomatic screening, family reproductive planning, carrier testing, prenatal diagnosis, and prophylactic intervention.
Currently, several groups describe the ethical obligation to return secondary findings (SFs) in the research setting; however, there remains substantial controversy and no standard accepted method for reporting.4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22 In fact, the program director of the National Human Genome Research Institute’s Ethical, Legal and Social Implications Research Program likened the topic to “arguably the most pressing issue in genetics today.”23 The debated issue within genomic research has been whether to follow the traditional researcher–participant model, where participants receive no feedback even when clinically urgent and actionable information is discovered or to go against convention and provide patients with the option to consent for the disclosure of SFs.4,6,8,15,16,17,19,21,24,25,26,27 One method for disclosure involves the use of a structured model to clinically analyze and categorize relevant secondary results through binning based on the degree of clinical utility and clinical validity.10,28 The ACMG recommends that “laboratories and clinics utilizing whole-genome sequencing/whole-exome sequencing should have clear policies in place related to disclosure of SFs. Patients should be informed of those policies and the types of SFs that will be reported back to them and under what circumstances. Patients should be given the option of not receiving certain SFs.”1 It has been questioned whether clinicians may have a legal obligation to release particular SFs; however, to date, there have been no legal cases related to clinical genomic testing.15 In a recent paper, the ACMG provided recommendations for the return of SFs, which involves the return of variants identified from a provided list of 56 genes without regard for the preference of the patient for these findings.29 Despite the vast and heated opinions of researchers, clinicians, and committees on the subject of SFs, few studies document the preferences of patients/research participants. Among the relatively few studies which do exist, participants in research studies express a desire for the option to receive results and disclosure decisions depend on a number of factors including the type, conclusiveness, and nature of the results.24,30,31,32 However, participants in research cohorts are generally healthy or facing non–life-threatening disease (e.g., refs. 3,30,31) and preferences for SFs may differ as compared with those of patients undergoing diagnostic exome sequencing (DES), who are generally affected with chronic and/or life-threatening illnesses.
Given the recent availability of DES, there is a limited knowledge regarding the patient preferences for receiving SFs in the clinical setting. On the basis of the unprecedented experience of a large reference laboratory providing clinical exome sequencing (Ambry Genetics, Aliso Viejo, CA), we provide an analysis of SFs reporting consent among the first 200 patients undergoing diagnostic whole-exome testing.
Materials and Methods
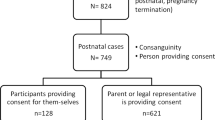
DNA from 200 patient families sent to Ambry Genetics was extracted from peripheral blood samples and, in rare cases, from processed DNA. Samples were prepared using the SureSelect Target Enrichment System (v2) (Agilent Technologies, Santa Clara, CA) and sequenced using paired-end, 100-cycle chemistry on the Illumina HiSeq 2000 (Illumina, San Diego, CA). A SFs reporting strategy was constructed based on recommendations regarding testing in minors from various professional organizations24,33,34,35 with which the majority of clinical genetics professionals agree,36 upon consultation with medical ethicists (H. Greely and M. Cho, personal communication), and was subsequently supported by the ACMG policy statement regarding the clinical application of genomic sequencing1 (Supplementary Table S1 online). Our laboratory requires genetic counseling and DES consenting to aid in informing patients of the complexity of exome sequencing and to allow them the option to consent to or decline SFs disclosure (http://ambrygen.com/requirements).
A parent or legal guardian (referred to collectively herein as “guardians”) completes the consent form for patients younger than the age of 18 years. Given that the impact and significance of SFs differs widely from that of primary results, patients are informed that SFs are reported separately from the primary exome report. The consent also details that reported SFs include only medically reviewed and previously defined mutations in characterized genes; variants of uncertain significance are not reported. At the time of consent, these SFs included four categories: (i) category A: carrier status of recessive disorders, (ii) category B: predisposition to later-onset disease, (iii) category C: predisposition to increased cancer risk, and (iv) category D: early-onset disease. Within the consent form, patients were provided with the option to receive a SFs report for each of the four categories. Adults (older than 18 years of age) chose among all four categories, whereas minors (younger than 18 years of age) chose whether to receive a category D result. A complete list of the genes and diseases from each of the four categories is available online (http://ambrygen.com/sites/default/files/pdfs/forms/most_updated/Incidental%20Findings_051313.pdf). The consent form contains example scenarios within each of the four categories, provided for the purpose of aiding, but not to take the place of, genetic counseling.
Patient demographics (age, diagnosis, and gender) and SFs disclosure decisions were collected from consent forms supplied to the laboratory with the requisition form and the biospecimens. Patient identifiers were removed. Retrospective data analysis of anonymized data exempted the study from the review of institutional review board. Data curation included the primary exome test option ordered, patient age, diagnosis and/or clinical description, and SFs disclosure elections among the four described categories. Statistical analyses were computed by χ2 goodness-of-fit tests and Fisher’s exact probability.
Results
The 200 probands were predominantly from the United States and Canada, and 98% (195/200) were referred by a geneticist/genetic counselor. The five cases which did not involve a geneticist or genetic counselor were ordered for probands younger than the age of 18 years. Moreover, 81% (n = 162) of the referred probands were children (younger than 18 years of age) ( Table 1 ). The average ages among children and adults were 5 and 38 years, respectively, with mean ages of 4 and 33 years, respectively. All patients were affected with a genetic disorder. The most common referral indications were neurological disease, multiple congenital anomalies, musculoskeletal anomalies, and metabolic disease (data not shown). Among the referrals within the adult cohort, common referrals also included cancer predisposition, immunologic disease, and nonsyndromic intellectual disability; metabolic disease was less common.
The majority of patients (187/200; 93.5%) consented to SFs reporting for one or more available categories ( Table 2 ). Guardians of 7 of 162 children (4%) did not consent to SFs reporting of category D, the one category available to them, whereas 16% (6/38) of adults did not consent to SFs reporting of one or more category, making adults significantly less likely to consent to SFs reporting (16 vs. 4%, respectively; P < 0.0001). Of the six adults who chose to blind one or more SFs, options for blinding were scattered between the four different categories, and only one chose to blind all four categories. Specifically, 8% (3/38) did not consent to SFs reporting of recessive disease carrier status; 11% (4/38) cancer predisposition, 8% (3/38) adult-onset disease predisposition, and 8% (3/38) early-onset disease. Among all patients regardless of age, 5% (10/200) did not consent to SFs reporting of early-onset disease. Lack of consent to SFs reporting among each category was evenly distributed between among six adult patients ( Table 3 ). Of note, only one adult did not consent to SFs reporting of all four categories. Overall, patients were significantly more likely to consent to SFs reporting than not (P < 0.0001).
Discussion
Overwhelmingly, patients undergoing DES consent to disclosure of all available SFs. The health status among the cohort of subjects herein emerges as a significant variable. Individuals pursuing DES may be more active in seeking medical information and therefore more receptive to obtaining SFs information than healthy individuals. Each of these factors may decrease fear regarding future disease risk while heightening desire to both find any possible contribution to a possible diagnosis as well as be prepared for additional findings which may further burden current health. In general, patients undergoing DES represent an extreme end of the health spectrum which introduces a substantial bias regarding threshold for disease risk acceptance, not likely generalizable to the general population.
Likelihood to consent is significantly greater among DES patients younger than the age of 18 years, perhaps further implicating health status as a factor influencing the impetus to opt for disclosure. The majority of patients within the child cohort were affected with complex diseases including severe cognitive impairment and a truncated life expectancy and represent the most severe end of the phenotypic spectrum. Most often, these children are faced with life-threatening illnesses with truncated life expectancies, have spent years or even decades in search of a diagnosis, and may have multiple affected family members. The adult DES patients had milder disease manifestations and were less likely affected with life-threatening diseases or severe cognitive impairment. However, the observed difference between preferences among adults and children might also reflect a difference between information parents would like to learn about their children, versus information that adult patients would like to learn about themselves.
No apparent sex or age bias emerged when comparing consenting versus nonconsenting adult DES patients. Of note, among the six adults opting for blinding of SFs, there is no trend toward blinding of any specific subcategory, possibly indicating an intricate and complicated decision-making process, more closely matching that of healthy participants in research studies where disclosure decisions depend on a number of factors, likely based on personal life experiences.24,30,31,32
It is curious that 15% of guardians opted for secondary reporting for predictive genetic test results including predisposition and carrier status for their children, despite the unavailability of these results to minors. It is possible that this result was confounded by practicalities such as the quality of genetic counseling, time constraints in busy clinics, or limitations in the understanding of the implications of these SFs. Although this may have been a mistake or oversight by the clinician or guardian at the time of consent, the choice for these findings draws attention to the issue of genetic testing in minors, which has been debated for years (e.g., refs. 24,32,33,34,35,36,37,38,39). Notably, the recent ACMG guidelines for SFs reporting recommend the disclosure of several adult-onset predisposition disease genes.29 In general, testing in minors has been discouraged when the benefits of the test will not accrue until adulthood such as with late-onset diseases or conditions without medical management or clinical significance.4 Genetic testing in minors is only recommended when the current potential benefits outweigh the harms.4,24 Those opposed to predictive testing believe that it may take away their autonomy, hinder their confidentiality, and believe the possibility of harm to be greater than the benefits of disclosing results to the parent/guardian.24,38 Those in favor of testing describe avoiding paternalism, increasing patient autonomy, and reducing anxiety among other psychological benefits.30,38 Notably, cognitive impairment has been discussed among the limited circumstances in which predictive genetic testing in minors may be appropriate.39 Studies have shown few patients have regretted receiving results for predictive genetic testing.36,37 Overall, a recent chronological review of the ethical arguments for predictive genetic testing in minors describes a gradual shift toward ethical arguments in favor of testing.38 Other studies cite this same trend.24
A potential source of variation in our data is in the level and depth of counseling/consenting provided to families referred for exome sequencing from clinicians with inherently different counseling styles, from clinics with varying levels of patient loads and processes. The majority of cases analyzed in this study involved a clinical geneticist or genetic counselor in the consent process, highlighting the quality of counseling. However, this potential variability in the delivery of counseling and consent did not appear to influence family decisions given that families overwhelmingly opted for SFs disclosure.
Overall, the data herein provide, for the first time, a preliminary study of the SFs report preferences among a small cohort of families undergoing DES in a clinical setting including 161 severely affected children and 39 somewhat less severely affected adults. Larger cohort studies including direct patient interviews to further define the implications and to address the impact of this strategy of SFs results return are warranted.
Disclosure
The authors of this manuscript are all employed and receive a salary from Ambry Genetics. Exome sequencing is among the commercially available tests.
References
ACMG Board of Directors. Points to consider in the clinical application of genomic sequencing. Gen Med 2012;14:759–761.
Bell CJ, Dinwiddie DL, Miller NA, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med 2011;3:65ra64.
Johnston JJ, Rubinstein WS, Facio FM, et al. Secondary variants in individuals undergoing exome sequencing: screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am J Hum Genet 2012;91:97–108.
Wolf SM, Lawrenz FP, Nelson CA, et al. Managing incidental findings in human subjects research: analysis and recommendations. J Law Med Ethics 2008;36:219–248, 211.
Wolf SM, Crock BN, Van Ness B, et al. Managing incidental findings and research results in genomic research involving biobanks and archived data sets. Genet Med 2012;14:361–384.
Wolf SM . The past, present, and future of the debate over return of research results and incidental findings. Genet Med 2012;14:355–357.
Van Ness B . Genomic research and incidental findings. J Law Med Ethics 2008;36:292–297, 212.
National Heart, Lung, and Blood Institute working group, Fabsitz RR, McGuire A, Sharp RR et al. Ethical and practical guidelines for reporting genetic research results to study participants: updated guidelines from a National Heart, LUng, and Blood Institute working group. Circ Cardiovasc Genet 2010;3:574–580.
Chahrour MH, Yu TW, Lim ET, et al.; ARRA Autism Sequencing Collaboration. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet 2012;8:e1002635.
Berg JS, Adams M, Nassar N, et al. An informatics approach to analyzing the incidentalome. Genet Med 2013;15:36–44.
Lohn Z, Adam S, Birch P, Townsend A, Friedman J . Genetics professionals’ perspectives on reporting incidental findings from clinical genome-wide sequencing. Am J Hum Genet 2012;999(Pt A):991–998.
Bredenoord AL, Kroes HY, Cuppen E, Parker M, van Delden JJ . Disclosure of individual genetic data to research participants: the debate reconsidered. Trends Genet 2011;27:41–47.
Pulley J, Clayton E, Bernard GR, Roden DM, Masys DR . Principles of human subjects protections applied in an opt-out, de-identified biobank. Clin Transl Sci 2010;3:42–48.
McGuire AL, Lupski JR . Personal genome research: what should the participant be told? Trends Genet 2010;26:199–201.
Clayton EW, Haga S, Kuszler P, Bane E, Shutske K, Burke W . Managing incidental genomic findings: legal obligations of clinicians. Genet Med 2013;15:624–629.
Bledsoe MJ, Clayton EW, McGuire AL, Grizzle WE, O’Rourke PP, Zeps N . Return of research results from genomic biobanks: a call for data. Genet Med 2013;15:159–160.
Bledsoe MJ, Clayton EW, McGuire AL, Grizzle WE, O’Rourke PP, Zeps N . Return of research results from genomic biobanks: cost matters. Genet Med 2013;15:103–105.
Bledsoe MJ, Grizzle WE, Clark BJ, Zeps N . Practical implementation issues and challenges for biobanks in the return of individual research results. Genet Med 2012;14:478–483.
Fullerton SM, Wolf WA, Brothers KB, et al. Return of individual research results from genome-wide association studies: experience of the Electronic Medical Records and Genomics (eMERGE) Network. Genet Med 2012;14:424–431.
Gliwa C, Berkman BE . Do researchers have an obligation to actively look for genetic incidental findings? Am J Bioeth 2013;13:32–42.
Clayton EW . Incidental findings in genetics research using archived DNA. J Law Med Ethics 2008;36:286–291, 212.
Lemke A, Bick D, Dimmock D, Simpson P, Veith R . Perspectives of clinical genetics professionals toward genome sequencing and incidental findings: a survey study. Clin Genet 2013;84:230–236.
Couzin-Frankel J . Human genome 10th anniversary. What would you do? Science 2011;331:662–665.
Abdul-Karim R, Berkman BE, Wendler D, et al. Disclosure of incidental findings from next-generation sequencing in pediatric genomic research. Pediatrics 2013;131:564–571.
Biesecker LG . The Nirvana fallacy and the return of results. Am J Bioeth 2013;13:43–44.
Biesecker LG . Secondary variants and human subjects research. Genet Med 2013;15:157.
Knoppers BM, Deschênes M, Zawati MH, Tassé AM . Population studies: return of research results and incidental findings Policy Statement. Eur J Hum Genet 2013;21:245–247.
Parker LS . The future of incidental findings: should they be viewed as benefits? J Law Med Ethics 2008;36:341–351, 213.
Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013;15:565–574.
Murphy J, Scott J, Kaufman D, Geller G, LeRoy L, Hudson K . Public expectations for return of results from large-cohort genetic research. Am J Bioeth 2008;8:36–43.
Kirschen MP, Jaworska A, Illes J . Subjects’ expectations in neuroimaging research. J Magn Reson Imaging 2006;23:205–209.
Wilfond BS, Carpenter KJ . Incidental findings in pediatric research. J Law Med Ethics 2008;36:332–340, 213.
Points to consider: ethical, legal, and psychosocial implications of genetic testing in children and adolescents. ASHG, ACMG board of directors. Am J Hum Genet 1995;57:1233–1241.
AMA code of medical ethics’ opinion on genetic testing. The virtual mentor. 2009;11:683–685.
Borry P, Evers-Kiebooms G, Cornel MC, Clarke A, Dierickx K . Genetic testing in asymptomatic minors: background considerations towards ESHG recommendations. Eur J Hum Genet 2009;17:711–719.
Duncan RE, Savulescu J, Gillam L, Williamson R, Delatycki MB . An international survey of predictive genetic testing in children for adult onset conditions. Genet Med 2005;7:390–396.
Duncan RE, Gillam L, Savulescu J, Williamson R, Rogers JG, Delatycki MB . “You’re one of us now”: young people describe their experiences of predictive genetic testing for Huntington disease (HD) and familial adenomatous polyposis (FAP). Am J Med Genet C Semin Med Genet 2008;148C:47–55.
Mand C, Gillam L, Delatycki MB, Duncan RE . Predictive genetic testing in minors for late-onset conditions: a chronological and analytical review of the ethical arguments. J Med Ethics 2012;38:519–524.
Ross LF, Ross LF, Saal HM, David KL, Anderson RR ; American Academy of Pediatrics; American College of Medical Genetics and Genomics. Technical report: ethical and policy issues in genetic testing and screening of children. Genet Med 2013;15:234–245.
Acknowledgements
We thank the genetic counselors and geneticists for providing fruitful feedback during the construction of the exome-sequencing patient consent form and for referring their patients to us for whole-exome sequencing. We would also thank Kamelia Shoaepour and Josh Keiles for their assistance with the organization of exome-sequencing patient consent forms. We also thank Hank Greely and Mildred Cho for their ethical consults during the development of the consent.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Table S1
(DOC 36 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Shahmirzadi, L., Chao, E., Palmaer, E. et al. Patient decisions for disclosure of secondary findings among the first 200 individuals undergoing clinical diagnostic exome sequencing. Genet Med 16, 395–399 (2014). https://doi.org/10.1038/gim.2013.153
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2013.153
Keywords
This article is cited by
-
Evaluation of a two-step model of opportunistic genomic screening
European Journal of Human Genetics (2024)
-
Parents, their children, whole exome sequencing and unsolicited findings: growing towards the child’s future autonomy
European Journal of Human Genetics (2021)
-
A qualitative study among patients with an inherited retinal disease on the meaning of genomic unsolicited findings
Scientific Reports (2021)
-
Disclosure of secondary findings in exome sequencing of 2480 Japanese cancer patients
Human Genetics (2021)
-
Exome sequencing in clinical settings: preferences and experiences of parents of children with rare diseases (SEQUAPRE study)
European Journal of Human Genetics (2019)
