
Abstract
Advances in genomics have near-term impact on diagnosis and management of monogenic disorders. For common complex diseases, the use of genomic information from multiple loci (polygenic model) is generally not useful for diagnosis and individual prediction. In principle, the polygenic model could be used along with other risk factors in stratified population screening to target interventions. For example, compared to age-based criterion for breast, colorectal, and prostate cancer screening, adding polygenic risk and family history holds promise for more efficient screening with earlier start and/or increased frequency of screening for segments of the population at higher absolute risk than an established screening threshold; and later start and/or decreased frequency of screening for segments of the population at lower risks. This approach, while promising, faces formidable challenges for building its evidence base and for its implementation in practice. Currently, it is unclear whether or not polygenic risk can contribute enough discrimination to make stratified screening worthwhile. Empirical data are lacking on population-based age-specific absolute risks combining genetic and non-genetic factors, on impact of polygenic risk genes on disease natural history, as well as information on comparative balance of benefits and harms of stratified interventions. Implementation challenges include difficulties in integration of this information in the current health-care system in the United States, the setting of appropriate risk thresholds, and ethical, legal, and social issues. In an era of direct-to-consumer availability of personal genomic information, the public health and health-care systems need to prepare for an evidence-based integration of this information into population screening.
Genet Med 2013:15(6):437–443
Similar content being viewed by others


Main
In this issue of Genetics in Medicine, Chowdhury et al.1 report on the recommendations of multidisciplinary expert workshops convened by the Foundation for Genomics and Population Health (PHG Foundation) in partnership with the University of Cambridge. Participants examined scientific, ethical, and logistical aspects of personalized population screening for prostate and breast cancer based on polygenic susceptibility. The authors recognized the promise of genetic stratification in population screening for breast and prostate cancer and identified key issues that need to be addressed before genetic stratification can be implemented in practice, the most important of which is the need to recognize the benefits and harms of stratified screening as compared with existing screening methods. They also identified several ethical issues such as discrimination of high-risk individuals and patient autonomy in relation to genetic testing of minors; the need for transparency and clear communication about genetic risk scores; and the need to develop new professional competencies and to assess cost effectiveness and acceptability of stratified screening programs before implementation.
We commend the authors for the thoughtful analysis and modeling of potential for improved effectiveness of screening and for early stakeholder engagement in a rapidly moving field. Here, we elaborate on the potential of population screening in an age of genomics and personalized medicine. At the outset, we acknowledge that although the promise of genomics will be first fulfilled in the diagnosis and management of monogenic disorders, its use in population screening for common complex diseases will lag behind due to significant evidentiary as well as implementation challenges.
Population Screening in the Age of Personalization
The idea of population screening of healthy individuals has been around for more than 100 years and has captured the interest of health-care providers, public health professionals, and the general public.2 The main purpose of screening is early detection of asymptomatic disease or risk assessment for future disease to improve health outcomes. Scientific and logistical principles for screening have been discussed by many organizations, most notably Wilson and Junger’s 1968 World Health Organization criteria,3 and have evolved over time.2 These principles ensure that the benefits of screening programs outweigh potential harms. Generally, people tend to overestimate the positive health impact of screening and underestimate the potential for harmful effects such as overdiagnosis, inappropriate interventions, and anxiety.4 For many years, the US Preventive Services Task Force (USPSTF) has regularly conducted systematic reviews of benefits and harms for screening programs and developed evidence-based recommendations.5
One of the criticisms leveled against evidence-based population screening is that guidelines typically apply to the “average” person in the population and may not be relevant to subgroups of the population with differing levels of risk, a fundamental tenet of personalized medicine that is largely driven by advances in genomics.6 For example, in breast cancer screening, the USPSTF currently recommends biennial screening mammography for women aged 50–74 years.7 However, they do acknowledge that the decision to start regular screening mammography before the age of 50 years should be an individual one and should take patient context into account, including the patient’s other risk factors and values regarding specific benefits and harms. This second recommendation reflects the continued debate and uncertainty about the value of breast cancer screening in those younger than 50.8 For prostate cancer, the USPSTF currently recommends against prostate-specific antigen screening in men, as a result of a recent systematic reviews of the balance of benefits and harms from such screening.9 The debate over prostate cancer screening in the United States has been ongoing for years, and the USPSTF recently concluded that the harms outweigh any potential benefits of screening for the majority of men.
Genetics and Population Screening
Could advances in genomics improve the benefits of population screening beyond “average risk” screening guidelines? Rapid progress in genomics is unraveling the genetic architecture of human disease.10 The applications of genetic discoveries for >2,500 monogenic diseases with available tests (such as Huntington disease and cystic fibrosis) have been relatively straightforward in terms of diagnosis and counseling of patients and relatives.11 Advances in whole-genome sequencing are promising near-term applications into the work-up of rare conditions strongly suspected to have a genetic basis.12 Already, a few applications of next-generation sequencing have been used in practice, and more is to come as the price of this technology declines and its analytic performance is enhanced.13
For almost 50 years, newborn screening has been the poster child of population screening as more and more countries around the world screen all newborns for an increasing number of rare genetic and metabolic conditions in order to prevent early death or disability.14 Screening tests are known to have high false-positive rates and thus are poorly predictive of disease for any given individual in the population. Screening tests are usually followed by more definitive diagnostic tests. In these settings, the emphasis of population screening is to find all cases of disease in the population (maximizing sensitivity). Individuals who are labeled as positive as a result of screening often undergo follow-up procedures, tests, or interventions and may experience anxiety. As a result, recent reviews of population-based screening have emphasized the need for a systematic evidence-based approach to assess both the benefits and harms of population screening to individuals and populations.15
In addition to newborn screening, a few selected monogenic disorders provide a window on personalization of screening recommendations in adults. For example, in the case of hereditary breast and ovarian cancer, the USPSTF currently recommends that all women with strong family history for breast and ovarian cancer be offered genetic counseling and evaluation for BRCA testing to reduce morbidity and mortality from breast and ovarian cancer.16 Generally, BRCA genes account for a small percent of breast cancer in the population but have a much higher absolute risk of disease among affected individuals. Similarly, the USPSTF recommends that colorectal cancer (CRC) screening is begun at the age of 50 years for people at an average risk in the population.17 However, for about 1 in 400 people, who have a highly penetrant single-gene disorder, Lynch syndrome (which accounts for 3–5% of all CRC), clinical guidelines recommend that CRC screening be started in patients in the twenties.18 The Evaluation of Genomic Applications in Practice and Prevention working group, an evidence panel sponsored by the Centers for Disease Control and Prevention that was modeled after the USPSTF in genomic medicine, recently recommended that all newly diagnosed cases of CRC in the population be tested for Lynch syndrome, cascade testing be done for their relatives to make early diagnosis of Lynch syndrome, and earlier and more frequent CRC screening and surveillance be instituted.19
Does Polygenic Inheritance Have a Role in Population Screening?
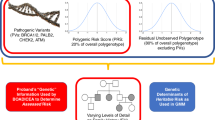
The contribution of genetics to most common diseases has long been captured under the rubric of polygenic inheritance,20 in which additive effects of numerous genes along with environmental determinants create a normal distribution of disease risk in the population. Until recently, the polygenic contribution to disease remained elusive and could only be described through metrics of heritability obtained from twin and family studies.21 A flurry of recent genome-wide association studies has changed the landscape, however, with hundreds of genetic variants uncovered for a wide variety of diseases.22
To explore the clinical validity and utility of polygenic information, studies have been conducted for type 2 diabetes, coronary heart disease, breast cancer, prostate cancer, multiple sclerosis, and others.23,24,25,26,27,28,29,30 Collectively, analyses show that the contribution of a combination of multiple alleles at multiple loci will be limited in predicting disease for any given individual. This is because the effect of individual variants on disease risk is quite modest (usually 5–20% increase in risk; odds ratios of 1.05–1.20), and thus most individuals are at slightly increased or slightly decreased risk as compared with the average risk of the underlying population. Even for people at the extremes of the distribution, disease risk is moderately increased, and very few people belong to the extremes of the distribution. For example, Pashayan and Pharoah31 show that for the 30 prostate cancer risk alleles, the 5% of the population at highest risk will have a twofold relative risk as compared with the population average. Furthermore, many of these studies show that adding polygenic information to risk-prediction models, when available, provide no or little additional discrimination (as reflected in analyses of the area under the curve) to current risk-prediction models based on traditional risk factors such as age, body mass index, and lipid levels. As discussed by Chowdhury et al.,1 a risk-stratification model must have adequate discrimination and calibration, and it should produce several strata of the population for which different management strategies are needed to improve population health outcomes. The clinical utility of testing for polygenic inheritance is established if testing provides better predictive ability than existing strategies, or comparable predictive ability at lower costs.
From what we know today, these conditions are largely unmet for most common human diseases. In fact, for most diseases, family history remains the most consistent risk factor, even after considering available genetic variants, perhaps reflecting unmeasured genes, shared environments, or complex gene–environment interactions.32 Collectively, the findings from genome-wide association studies do not explain the known familial clustering of common diseases,33 which is also likely due to shared environmental factors and complex gene–environment interactions. Thus, risk profiles obtained from known genetic variants do not provide sufficient discrimination to warrant integration of polygenic inheritance in individual disease prediction. As Wald and Morris34 comment in a recent paper, “there is little scope for genetic testing in the prediction of common disease; it is an area where hope unfortunately trumps the negative evidence. Risk factors that can make a significant contribution to the burden of a disease are, within a population, usually too weakly associated with the disease they cause to be useful predictors of who will become affected. Common diseases occur commonly, and it is usually not useful to screen for something that is common. In such circumstances, a population-wide approach is needed that is simple, effective, and safe. Screening based on age alone may be enough in these circumstances; the use of more complex assessments can be a distraction and unfruitful.”34
Given that polygenic inheritance leads to low individual risk prediction and personalized interventions, could it still be useful as a risk stratifier in population screening to target population subgroups at high risk of disease or even find subgroups at low risk of disease to avoid testing and other interventions that carry both potential benefits and harms? Pashayan and Pharoah31 have previously pointed to the potential for clinical utility of polygenic risk stratification in population-based screening for breast cancer. In the United Kingdom, the National Health Service offers screening to women between the ages of 47 and 69 years (47–69 from 2012). This age-based criterion for eligibility to screening is suboptimal because many women younger than 50 years will develop cancer and most women older than 69 years will not develop breast cancer. However, identical absolute risk thresholds for screening could also be determined by a combination of age and polygenic risk profile. Under this schema, they compared breast cancer screening in which women are screened from the age of 47 (10-year absolute risk of ≥2%), and genetically stratified screening in which women are screened at 2.5% absolute risk calculated on the basis of age and polygenic risk. Using an age-based criterion alone, 65% of women would be eligible for screening, with 85% of cases detected in the screened population. Using a stratified screening strategy, 50% of women would be screened, with 73% of cases detected. Thus, the number of women eligible for screening would be 24% fewer at a cost of 14% fewer screen-detectable cases.31 Assuming all possible genetic susceptibility variants for breast cancer are known, they found that 28% would be screened and 76% of the cases would occur in the screened population. As compared with screening from age 47, 57% fewer women would be screened at a cost of detecting 10% fewer cases. Thus, “a reduction in the number of individuals offered screening would also reduce the harms associated with screening, including a reduction in overdiagnosis and a reduction in false-positives with all the consequences that these adverse outcomes entail.”31 From our perspective, for a not-too-realistic scenario, these numbers provide a theoretical upper bound for the role of polygenes in breast cancer as the remaining “missing heritability” may involve many rare genetic variations, complex gene–environment interactions, or posttranslational alterations such as epigenetics.
Nevertheless, this kind of reasoning, if supported by empirical data, could have a major influence on the way we think about population screening. For example, it would imply that some women currently getting screening would be missed, even if at better “efficiency” of the screening program. As discussed by Chowdhury et al.,1 one option is to retain current age categories and add younger groups with higher risk (which is something we currently do in the case of the very high risk associated with BRCA mutations). Another option is to use “absolute risk” as the main criterion for screening and start to exclude older women who are getting screened now. These issues are important when considering the overall impact of breast cancer screening. A recent analysis of US breast cancer surveillance data from 1976 to 2008 showed that despite substantial increases in the number of cases of early-stage breast cancer, screening has only marginally reduced the rate of advanced cancer. This suggests that that there is substantial overdiagnosis, and that screening is having, at best, only a small effect on the rate of death from breast cancer.35 Thus, it will be even more important for any stratified or personalized screening regimen that involves genetic factors to detect early cases of breast cancer that will benefit from screening (i.e., they have different natural history and are treatable), rather than purely using numerical thresholds of absolute risks. Currently, we have limited information on the impact of polygenic risk on breast cancer natural history.
Using modeling of available genetic variants in prostate cancer, the same authors have also explored genetic risk stratification in prostate cancer screening.36 They found that a stratified approach for screening could halve the number of screenings required and could reduce the overall costs associated with screening. However, even if we get a more complete picture of the polygenic inheritance in prostate cancer, current evidence in the United States shows more harms than benefits for prostate-specific antigen screening,9 especially for high-risk groups.37 This suggests that in prostate cancer, using polygenic profiles for risk assessment may not be as fruitful as for breast cancer. This is reflected in the recent Evaluation of Genomic Applications in Practice and Prevention–sponsored evidence review on the use of single-nucleotide polymorphism profiles in prostate cancer risk assessment.38 Again, issues of absolute risk thresholds for screening in relation to overdiagnosis and natural history apply to prostate cancer screening, similar to breast cancer screening as discussed above.
A third example, largely unexplored by the authors, is CRC screening. The USPSTF currently recommends screening for CRC using fecal occult blood testing, sigmoidoscopy, or colonoscopy in adults, beginning at the age of 50 years and continuing until 75 years.17 CRC screening, as with any screening, is most effective when it is applied to a large percentage of eligible people and utilized appropriately. In 2010, a National Institutes of Health state-of-the-science conference analyzed suboptimal national screening rates for CRC, identified the barriers to screening, and proposed solutions to increase screening rates.39 The independent panel found that despite substantial progress toward higher CRC screening rates nationally, screening rates fall short of desirable levels. Targeted initiatives to improve screening rates and reduce disparities in underscreened communities and population subgroups could further reduce CRC morbidity and mortality. Could targeting on the basis of genetic susceptibility improve the overall effectiveness of CRC screening? As with breast cancer, an increasing number of genetic variants with weak additive effects have been associated with an increased risk of CRC.40 This raises the possibility that some population stratification of risk for CRC, on the basis of polygenic inheritance, can be undertaken in conjunction with age and family history to improve the uptake and efficiency of current screening. It has long been known that individuals with first-degree relatives with CRC have an almost twofold increase in the risk of CRC.41 For these individuals, their absolute risk of CRC is almost the same at 40 years as compared with an “average” 50-year-old person drawn from the population.42 Recently, Wilschut et al.43 evaluated how many screenings should be recommended to individuals with various degrees of family history. On the basis of population data, they conducted modeling to estimate the impact of stratified CRC screening strategies varying by age at which screening is started and stopped in addition to the screening interval. They found that optimal screening strategies varied considerably with the number of affected first-degree relatives and their age of diagnosis. Shorter screening intervals than the currently recommended 5 years were found to be appropriate for the highest-risk group.43 With additional information, one would be able to do the same type of calculations that Pashayan and Pharoah did for breast cancer screening. Nonetheless, we could envision that in the case of CRC screening, polygenic inheritance will also provide a fuller distribution of CRC risk than family history alone, which includes people at the lower end of risks, who may or may not benefit from the recommended start of CRC screening at 50 years of age if their absolute risks are much lower than the population average, or may not need the same screening interval (10 years).
The Bottom Line: Need for Empirical Evidence
The use of polygenic risk information in population screening is an intriguing idea that needs to be further explored using empirical evidence, in addition to modeling. As discussed by Chowdhury et al.,1 and expanded below, many questions need to be answered before polygenic inheritance can be integrated into a population screening paradigm. First, the analytic validity of the polygenic test is an essential prerequisite for its use in practice for any clinical indication. Currently, the analytic performance of whole-genome analysis is rapidly improving with high sensitivity and specificity, but a relatively small error rate can translate into millions of false calls across the genome.44 Applications of polygenes will still have to wait for more reliable technology with quality control and assurance. Second, credible epidemiological data should be available on genetic and nongenetic risk factors in the population to be tested. This is where the information is so much in flux right now, and much of it is derived from case–control studies45 in which potential biases and limitations can interfere with inference and interpretation. These include the lack of representation of the spectrum of disease in cases, the potential nonrepresentativeness of controls, issues in confounding, selection bias and population stratification, and lack of consideration and/or accurate measurement of important nongenetic risk factors for the disease in question.46 The lack of reliable epidemiological information is compounded by the inability of current studies to address all common and rare genetic variations in the genome, the so-called missing heritability. Integration of whole-genome sequencing technology into well-powered large-scale population cohort studies will help fill the knowledge gaps in the next decade.47
Third, the computation of an overall age-specific absolute risk of disease conferred by multiple genetic variants will remain a major challenge for the foreseeable future.47 The current approach is to multiply the odds ratios associated with each risk variant to arrive at a composite risk score. This method of integration assumes statistical independence between risk variants and ignores the potential for undetected gene–environment interactions that alter risk, a scenario unlikely to endure as the number of discovered variants increases, potentially leading to overestimation of disease risk. One common approach is to simply sum up the number of well-replicated risk alleles for a specific disease that any given individual carries. Integrating risk conferred by multiple variants and nongenetic factors is probably the most daunting challenge in this regard. Due to the complex genetic architecture of many common diseases, it will be difficult to identify the dependencies and interactions among genetic and nongenetic risk factors. Given the large role of environmental factors in the development of common diseases, analytic integration of genetic and nongenetic factors to disease risk is crucial.48
Fourth, the impact of polygenic inheritance on the natural history of the disease needs to be understood. One of the major challenges of screening has been the need to identify individuals who will actually benefit from early detection: those who have aggressive forms of the disease that will need earlier interventions as opposed to indolent cases that could be diagnosed later. At this point, it is unclear whether polygenic risk stratification will aid in the differentiation between life-threatening cancers and mild, chronic forms (e.g., prostate cancer49;49 rapid progression of polyps in CRC50).
Fifth, the balance of benefits and harms of available interventions, including costs, to individuals and populations should be carefully studied when polygenic testing is considered to stratify risk. In this context, the ideal designs for evaluation are randomized clinical trials comparing the outcomes of risk stratification with current (nonstratified) practice.51 A crucial question to address is whether the interventions work the same way or have the same effects on disease natural history across the polygenic risk distribution. In this situation, randomized trials are quite expensive, require large sample sizes, and take long periods to implement. Observational, quasiexperimental, and comparative effectiveness studies have been recently proposed as additional tools in the evaluation of population stratification.52 The use of decision analysis and economic modeling can provide valuable additional information to inform decisions about pilot implementation studies. Such studies can collect real-world data on benefits, harms, and costs.53 Further methodological work is clearly needed in this field.
Sixth, it is important to consider the acceptability of this approach within the larger societal and policy issues54 in the United States. In a time of limited health-care resources in which there are major disparities in access to recommended preventive services to the population, would a polygenic risk stratification be viewed more or less favorably than other forms of stratification, for instance by age, income, or other population subgroups that are known to have differential rates of morbidity and mortality from the disease in question, as well as access to available interventions? Because the intervention has both clinical benefits to persons who are identified as positive through the screening and potential significant harms, such as anxiety, unnecessary health-care costs, and potentially harmful interventions (e.g., prostate cancer), it may make more sense to do risk stratification to maximize benefits and minimize harms. However, genetics is only one form of stratification, and we should not abide by the notion of “genetic exceptionalism” in this regard.55 We need to evaluate whether the polygenic information combined with other nongenetic risk factors can provide a rationale for a stratified approach to population screening as compared with current approaches that are geared to the “average” population. Certainly, stratification based on age (e.g., in cancer screening), although not a perfect discriminator, is much cheaper and easier to implement than stratification on the basis of whole-genome sequencing. Adding a few more variables could provide additional discrimination and may be closer to the clinical endpoints (e.g., body mass index, fasting glucose level in the prediction of type 2 diabetes). Nevertheless, one advantage for polygenic risks is that we can obtain this information at any point in life, predating by years or even decades the presence or absence of the endophenotypes (e.g., lipid levels), which occur closer to the onset of disease. If there is a rationale and scientific evidence that earlier intervention will make a difference on health outcomes, this will make a stronger case for using polygenic information to stratify population risks. Perhaps the biggest potential use of polygenic inheritance in population stratification may be the identification of low-risk groups that may require no or less screening. The symmetry of the polygenic curve allows risk stratification of additive genetic factors at both ends of the distribution, whereas most single-gene disorders like BRCA and Lynch syndrome give us only the upper end of risk, in which more or earlier interventions are needed. However, the acceptability of doing less screening for some segments of the population is uncertain and will need to be carefully explored.
How Does Polygenic Inheritance Fit With Direct-to-Consumer Personal Genomic Tests?
Finally, although our focus here is on “population utility” in the use of polygenic risk information, there is an emerging literature describing and quantifying the concept of “personal utility” of genomic risk information.56 This interest has evolved primarily as a result of the availability of personal genomic tests that are sold directly to consumers with or without the involvement of health-care providers.57 Information thus far available from personal genomic tests essentially derives from polygenic risk information obtained from genome-wide association studies. Varying opinions exist within the scientific community and the general public about the inherent value of genetic information for different purposes.58 We do not address these issues here and refer readers to previous publications on this topic including the need for a multidisciplinary research agenda, with a strong emphasis on behavioral, social, and communication sciences.59 It is crucial to engage consumers in shared decision making about the use of polygenic risk information for improving health. With dramatic improvements in genomic-sequencing technologies and expected drop in prices in the next few years, it is imperative for appropriate research to be conducted to allow scientific inferences on the value of polygenic risk information to individuals and populations before widespread integration of such information in clinical practice.
Concluding Remarks
Population screening for rare genetic diseases with high penetrance will continue to be a mainstay for genetic screening. However, technological developments will drive the interest in using polygenic inheritance as a risk stratifier in population screening for common disease. As discussed in this article and by Chowdhury et al.,1 this is an intriguing concept that has many evidentiary and implementation challenges. The incremental value of using polygenic inheritance in population screening, as compared with other forms of stratification, will have to be rigorously explored. In the meantime, full engagement of the scientific community, clinical and public health practice, consumers, and policy makers is required to prepare for the evidence-based integration of genomic information into health care and public health practice.
Disclosure
The authors declare no conflict of interest.
References
Chowdhury S, Dent T, Pashayan N, et al. Incorporating genomics in breast and prostate cancer screening: assessing the implications. Genet Med 2013, e-pub ahead of print February 14.
Harris R, Sawaya GF, Moyer VA, Calonge N . Reconsidering the criteria for evaluating proposed screening programs: reflections from 4 current and former members of the U.S. Preventive Services Task Force. Epidemiol Rev 2011;33:20–35.
Wilson JMG, Jungner G . Principles and Practice of Screening for Disease. World Health Organization: Geneva, Switzerland, 1968.
Harris R . Overview of screening: where we are and where we may be headed. Epidemiol Rev 2011;33:1–6.
United States Preventive Services Task Force. http://www.uspreventiveservicestaskforce.org/index.html Accessed 18 November 2012.
Hood L, Friend SH . Predictive, personalized, preventive, participatory (P4) cancer medicine. Nat Rev Clin Oncol 2011;8:184–187.
US Preventive Services Task Force. Screening for breast cancer: USPSTF recommendation statement. Ann Int Med 2010;151:716–726.
American College of Radiology: detailed ACR statement on ill advised and dangerous USPSTF mammography recommendations. http://gm.acr.org/MainMenuCategories/media_room/FeaturedCategories/PressReleases/UPSTFDetails.aspx Accessed 26 July 2012.
Moyer VA ; U.S. Preventive Services Task Force. Screening for prostate cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2012;157:120–134.
Hindorff LA, Gillanders EM, Manolio TA . Genetic architecture of cancer and other complex diseases: lessons learned and future directions. Carcinogenesis 2011;32:945–954.
National Center for Biotechnology Information: Genetic Testing Registry. http://www.ncbi.nlm.nih.gov/gtr/ Accessed 26 July 2012.
Bick D, Dimmock D . Whole exome and whole genome sequencing. Curr Opin Pediatr 2011;23:594–600.
Manolio TA, Green ED . Genomics reaches the clinic: from basic discoveries to clinical impact. Cell 2011;147:14–16.
National Newborn Screening and Genetics Resource Center. http://genes-r-us.uthscsa.edu/ Accessed 19 November 2012.
Gurian EA, Kinnamon DD, Henry JJ, Waisbren SE . Expanded newborn screening for biochemical disorders: the effect of a false-positive result. Pediatrics 2006;117:1915–1921.
US Preventive Services Task Force. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: recommendation statement. Ann Int Med 2005;143:355–361.
U S Preventive Services Task Force. Screening for colorectal cancer: recommendation statement. Ann Int Med 2008;149:627–637.
Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007;44:353–362.
Evaluation of Genomic Applications in Practice and Prevention (EGAPP) working group. Recommendation from the EGAPP working group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med 2009;11:35–41.
Lange K . An approximate model of polygenic inheritance. Genetics 1997;147:1423–1430.
Visscher PM, Hill WG, Wray NR . Heritability in the genomics era–concepts and misconceptions. Nat Rev Genet 2008;9:255–266.
Yu W, Yesupriya A, Wulf A, et al. GWAS Integrator: a bioinformatics tool to explore human genetic associations reported in published genome-wide association studies. Eur J Hum Genet 2011;19:1095–1099.
Willems SM, Mihaescu R, Sijbrands EJ, van Duijn CM, Janssens AC . A methodological perspective on genetic risk prediction studies in type 2 diabetes: recommendations for future research. Curr Diab Rep 2011;11:511–518.
Mihaescu R, Meigs J, Sijbrands E, et al. Genetic risk profiling for prediction of type 2 diabetes. PLoS Curr 2011;11:3RRN1208.
Kathiresan S, Melander O, Anevski D, et al. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med 2008;358:1240–1249.
Gail MH . Discriminatory accuracy from single-nucleotide polymorphisms in models to predict breast cancer risk. J Natl Cancer Inst 2008;100:1037–1041.
Wacholder S, Hartge P, Prentice R, et al. Performance of common genetic variants in breast-cancer risk models. N Engl J Med 2010;362:986–993.
Zheng SL, Sun J, Wiklund F, et al. Cumulative association of five genetic variants with prostate cancer. N Engl J Med 2008;358:910–919.
Salinas CA, Koopmeiners JS, Kwon EM, et al. Clinical utility of five genetic variants for predicting prostate cancer risk and mortality. Prostate 2009;69:363–372.
Jafari N, Broer L, van Duijn CM, Janssens AC, Hintzen RQ . Perspectives on the use of multiple sclerosis risk genes for prediction. PLoS ONE 2011;6:e26493.
Pashayan N, Pharoah P . Translating genomics into improved population screening: hype or hope? Hum Genet 2011;130:19–21.
Doerr M, Teng K . Family history: still relevant in the genomics era. Cleve Clin J Med 2012;79:331–336.
Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature 2009;461:747–753.
Wald NJ, Morris JK . Personalized medicine: hope or hype. Eur Heart J 2012;33:1553–1554.
Bleyer A, Welch HG . Effect of three decades of screening mammography on breast-cancer incidence. N Engl J Med 2012;367:1998–2005.
Kmietowicz Z . Adding age and genetic risk to PSA test could improve screening for prostate cancer. BMJ 2012;345:e7467.
Howard K, Barratt A, Mann GJ, Patel MI . A model of prostate-specific antigen screening outcomes for low- to high-risk men: information to support informed choices. Arch Intern Med 2009;169:1603–1610.
Agency for Healthcare Research Quality, technical report: multigene panels in prostate cancer risk assessment. http://effectivehealthcare.ahrq.gov/index.cfm/search-for-guides-reviews-and-reports/?pageaction=displayproduct&productid=1172 Accessed 19 November 2012.
NIH consensus development program. Enhancing the use and quality of colorectal cancer screening: final panel statement 2009. http://consensus.nih.gov/2010/colorectalstatement.htm Accessed 26 July 2012.
Hawken SJ, Greenwood CM, Hudson TJ, et al. The utility and predictive value of combinations of low penetrance genes for screening and risk prediction of colorectal cancer. Hum Genet 2010;128:89–101.
Butterworth AS, Higgins JP, Pharoah P . Relative and absolute risk of colorectal cancer for individuals with a family history: a meta-analysis. Eur J Cancer 2006;42:216–227.
Fuchs CS, Giovannucci EL, Colditz GA, Hunter DJ, Speizer FE, Willett WC . A prospective study of family history and the risk of colorectal cancer. N Engl J Med 1994;331:1669–1674.
Wilschut JA, Steyerberg EW, van Leerdam ME, Lansdorp-Vogelaar I, Habbema JD, van Ballegooijen M . How much colonoscopy screening should be recommended to individuals with various degrees of family history of colorectal cancer? Cancer 2011;117:4166–4174.
Ku CS, Wu M, Cooper DN, et al. Technological advances in DNA sequence enrichment and sequencing for germline genetic diagnosis. Expert Rev Mol Diagn 2012;12:159–173.
Khoury MJ, Gwinn M, Clyne M, Yu W . Genetic epidemiology with a capital E, ten years after. Genet Epidemiol 2011;35:845–852.
Manolio TA, Bailey-Wilson JE, Collins FS . Genes, environment and the value of prospective cohort studies. Nat Rev Genet 2006;7:812–820.
Manolio TA, Weis BK, Cowie CC, et al. New models for large prospective studies: is there a better way? Am J Epidemiol 2012;175:859–866.
Salari K, Watkins H, Ashley EA . Personalized medicine: hope or hype? Eur Heart J 2012;33:1564–1570.
Etzioni R, Gulati R, Tsodikov A, et al. The prostate cancer conundrum revisited: treatment changes and prostate cancer mortality declines. Cancer 2012;118:5955–5963.
Ransohoff DF, Pignone M, Russell LB . Using models to make policy: an inflection point? Med Decis Making 2011;31:527–529.
Teutsch SM, Bradley LA, Palomaki GE, et al. The Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Initiative: methods of the EGAPP working group. Genet Med 2009;11:3–14.
Goddard KA, Knaus WA, Whitlock E, et al. Building the evidence base for decision making in cancer genomic medicine using comparative effectiveness research. Genet Med 2012;14:633–642.
Veenstra DL, Roth JA, Garrison LP Jr, Ramsey SD, Burke W . A formal risk-benefit framework for genomic tests: facilitating the appropriate translation of genomics into clinical practice. Genet Med 2010;12:686–693.
Burke W, Tarini B, Press NA, Evans JP . Genetic screening. Epidemiol Rev 2011;33:148–164.
Evans JP, Burke W, Khoury M . The rules remain the same for genomic medicine: the case against “reverse genetic exceptionalism.” Genet Med 2010;12:342–343.
Grosse SD, McBride CM, Evans JP, Khoury MJ . Personal utility and genomic information: look before you leap. Genet Med 2009;11:575–576.
Myers MF, Bernhardt BA . Direct-to-consumer genetic testing: introduction to the special issue. J Genet Couns 2012;21:357–360.
Spencer DH, Lockwood C, Topol E, et al. Direct-to-consumer genetic testing: reliable or risky? Clin Chem 2011;57:1641–1644.
Khoury MJ, McBride CM, Schully SD, et al. The Scientific Foundation for Personal Genomics: recommendations from a National Institutes of Health-Centers for Disease Control and Prevention multidisciplinary workshop. Genet Med 2009;11:559–567.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Khoury, M., Janssens, A. & Ransohoff, D. How can polygenic inheritance be used in population screening for common diseases?. Genet Med 15, 437–443 (2013). https://doi.org/10.1038/gim.2012.182
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2012.182
Keywords
This article is cited by
-
Genetic profiling of decreased bone mineral density in an independent sample of Caucasian women
Osteoporosis International (2018)
-
Primary care providers’ cancer genetic testing-related knowledge, attitudes, and communication behaviors: A systematic review and research agenda
Journal of General Internal Medicine (2017)
-
Counselees’ Perspectives of Genomic Counseling Following Online Receipt of Multiple Actionable Complex Disease and Pharmacogenomic Results: a Qualitative Research Study
Journal of Genetic Counseling (2017)
-
Developing and evaluating polygenic risk prediction models for stratified disease prevention
Nature Reviews Genetics (2016)
-
Maximising the efficiency of clinical screening programmes: balancing predictive genetic testing with a right not to know
European Journal of Human Genetics (2015)
