
Abstract
Purpose:
Recent developments in genomics have led to expanded carrier screening panels capable of assessing hundreds of causal mutations for genetic disease. This new technology enables simultaneous measurement of carrier frequencies for many diseases. As the resultant rank-ordering of carrier frequencies impacts the design and prioritization of screening programs, the accuracy of this ranking is a public health concern.
Methods:
A total of 23,453 individuals from many obstetric, genetics, and infertility clinics were referred for routine recessive disease carrier screening. Multiplex carrier screening was performed and results were aggregated for this study.
Results:
Twenty-four percent of individuals were identified as carriers for at least one of 108 disorders, and 5.2% were carriers for multiple disorders. We report tabulations of carrier frequency by self-identified ethnicity and disease.
Conclusion:
To our knowledge, this study of a large, ethnically diverse clinical sample provides the most accurate measurements to date of carrier frequencies for hundreds of recessive alleles. The study also yields information on the clinical considerations associated with routine use of expanded panels and provides support for a pan-ethnic screening paradigm that minimizes the use of “racial” categories by the physician, as recommended by recent guidelines.
Genet Med 2013:15(3):178–186
Similar content being viewed by others


Introduction
Carrier screening is the practice of diagnosing individuals and couples at risk of conceiving children affected by recessive diseases. This practice is widespread and growing in importance, as carrier couples identified before conception may decide to pursue a variety of preventive measures, including in vitro fertilization with preimplantation genetic diagnosis, use of donor gametes, adoption, or early prenatal diagnostic testing.
In 2001 cystic fibrosis (CF) became the first disease recommended for routine carrier screening in the United States when the American Congress of Obstetrics and Gynecology (ACOG) and the American College of Medical Genetics (ACMG) jointly recommended that CF screening be offered to all individuals of Caucasian and Ashkenazi Jewish ethnicity and that it be “made available” to individuals of all other ethnic groups.1 In 2005, in recognition of increasing population ethnic admixture, the ACOG Committee on Genetics stated it was reasonable to offer CF screening pan-ethnically.2
Routine carrier screening for spinal muscular atrophy (SMA) has become more common since 2008 when ACMG recommended that screening be offered pan-ethnically.3 However, SMA screening is still not performed as routinely as CF testing,4,5 in part because of a conflicting ACOG statement that SMA should not be offered to the general population.6
In the absence of family history, carrier screening for diseases beyond CF and SMA is typically offered based on stated ethnicity. For example, hemoglobinopathy carrier screening is offered to individuals of African, Southeast Asian, or Mediterranean ancestry,7 whereas a panel of 4–16 diseases is routinely offered to individuals of Ashkenazi Jewish ancestry.8,9,10
However, the advent of modern genomics has changed the health economic calculus around genetic screening, with costs-per-megabase for DNA sequencing falling at a rate faster than Moore’s Law would indicate.11 Consequently, the amortized cost of carrier screening across the population for hundreds of genetic diseases is now far less than the cost of treating an affected child.12,13,14 Genomics has also called into question the reification of “racial” categories in clinical guidelines, as high-throughput assays now allow a physician to prescribe the same test for every patient, regardless of ethnicity, with little or no increase in cost.
Here we present clinical data from a large-scale deployment of expanded carrier screening across dozens of practices. In addition to providing perhaps the best estimates to date of a wide range of Mendelian allele frequencies, the deployment provides a concrete example of how genomics may reduce the role of “race” in biomedicine. It also identifies a number of severe, frequent, and easily detected disease alleles that have been omitted to date from population screening programs but that are easily included in expanded carrier testing panels.
Materials and Methods
Multiplex platform
The screening platform (Universal Genetic Test, Counsyl, South San Francisco, CA) uses high-throughput genotyping to identify disease-causing variants and corresponding wild-type alleles. In total, 417 disease-causing mutations associated with 108 recessive diseases were assayed and interpreted via fluorescent sequences. We consider a subset (listed in Supplementary Table S1 online) of the diseases identifiable via the platform. The analytical performance has been reported previously and compares favorably with traditional single-gene testing methods.12 Our reported disease carrier frequencies are lower bounds, as there are variants for each disease that the assay platform does not test.
Study population
This study sample included 23,453 individuals. Referral sources included family practitioners, geneticists, genetic counselors, obstetricians, perinatologists, and reproductive endocrinologists. Documented informed consent, including consent to research, was required of all patients and is on record at our facility. Institutional review board exemption is applicable due to de-identification of the data presented (45 CFR part 46.101(b)(4)). Genetic counseling was made available at no cost to all tested individuals.
Funding was performed as fee-for-service, typically reimbursed through third-party payers.
Disease panels
Diseases identifiable via the test platform range in severity from mild to incompatible with life. Nearly every disease can be considered severe (associated with infant or child mortality), significant (associated with progressive disease and reduced life span), or requiring significant intervention or treatment. The remainder is composed of milder conditions characterized by high allele frequencies and reduced penetrances, e.g., factor V Leiden thrombophilia (OMIM 188055), glucose-6-phosphate dehydrogenase deficiency (OMIM 305900), HFE-associated hereditary hemochromatosis (OMIM 235200), prothombin thrombophilia (OMIM 176930), pseudocholinesterase deficiency (OMIM 177400), and two common MTHFR mutations (OMIM 607093).
As the testing platform permits customization of disease panels, some physicians chose to prescribe testing for a subset of the full 417-mutation panel. In addition, ongoing improvements to the platform resulted in additions and refinements of various disease mutations. The sum effect of these changes is that the number of individuals tested for a given mutation varies moderately, as shown in the first column of Supplementary Table S1 online. All OMIM numbers are also noted in Supplementary Table S1 online.
Results
Data were available from 23,453 screened patients. Routine screening for a possible carrier state was the indication in all individuals. Screening for other indications, including potential gamete donor, infertility, and personal or family history, was also performed. Individuals with those indications were excluded from this study.
Population demographics
Self-reported ethnicity is summarized in Table 1 . Caucasians constituted 60.6%, and 75.0% were female. Most samples came from within the United States; a minority came from Australia, Canada, England, or New Zealand.
The median age was 33.0 years and mean age was 33.63 years. Most (n = 17,865) of the individuals were from 21–39 years of age. A small percentage was under 21 years (1.6%) or over 45 years (2.8%).
Disease carrier states
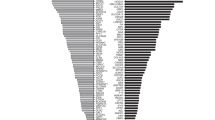
Alleles associated with 96 recessive diseases (excluding mild conditions) were identified in our 23,453 patients ( Supplementary Table S1 online). Among the mild conditions, the most common was MTHFR deficiency, with a frequency of 1 in 1.9. Because these conditions typically have limited reproductive decision-making significance, they will not be considered further in this analysis. All subsequent analysis considers only those diseases listed in Supplementary Table S1 online.
Of the total sample, 24.0% of individuals (n = 5,633) were heterozygous for at least one non-mild condition. A total of 7,067 heterozygous states were identified. Carrier statistics are fully reported in Supplementary Table S1 online.
Seventy-eight individuals were identified as homozygotes or compound heterozygotes for the following conditions: α-1-antitrypsin deficiency (n = 38), cystic fibrosis (n = 9), GJB2-related DFNB1 nonsyndromic hearing loss and deafness (n = 6), factor XI deficiency (n = 5), Gaucher disease (n = 4), familial Mediterranean fever (n = 3), carnitine palmitoyltransferase II deficiency (n = 2), medium chain acyl-CoA dehydrogenase deficiency (n = 2), sickle cell disease (n = 2), short chain acyl-CoA dehydrogenase deficiency (n = 2), achromatopsia (n = 1), β-thalassemia (n = 1), hexosaminidase A deficiency (n = 1), familial dysautonomia (n = 1), lipoamide dehydrogenase deficiency (n = 1), Niemann–Pick disease type C (n = 1), Pompe disease (n = 1), and spinal muscular atrophy (n = 1). The published specificity of the testing method suggests that these are “true positives” and merit further examination of clinical correlations.12 Review of clinical notes found at least two individuals with previously known diagnoses, for Gaucher disease and deafness. Another individual reported a history of sickle cell disease but did not specify whether this was familial or personal. All others did not report diagnosis information in their test requisitions and selected the routine carrier screening indication. For asymptomatic individuals with conditions such as α-1-antitrypsin deficiency, results could be used to guide appropriate surveillance.
Carrier rate variability by ethnicity
Frequencies of positive results varied by ethnic group ( Table 1 ). On average, 24.0% of individuals were positive for at least one condition. When stratified by self-reported ethnicity, this frequency ranged from 43.6% of Ashkenazi Jewish individuals to 8.5% of East Asians. For ethnic groups like the Ashkenazi Jewish, this frequency is unsurprising given the availability of screening for many conditions found in this group.
Multiple-disease carriers
Some individuals were heterozygous for multiple disorders ( Table 2 ), with ~5.2% (n = 1,210) found to be carriers of two or more disorders. Most were heterozygous for only two conditions (4.3% of all screenees and 83.9% of multiple-disease carriers), although a small number were carriers of three (0.7 and 13.8%) or more than three conditions (0.1 and 2.3%). Ashkenazi Jewish individuals were most frequently identified as multiple carriers, with 13.3% of all tested Ashkenazi Jews carrying more than one genetic disorder. These values are unsurprising, as population geneticists have long known that individuals carry on average 4–5 recessive lethal alleles.10,15 Put another way, the average number of positive results for recessive lethals in this panel is only (0 × 0.694 + 1 × 0.244 + 2 × 0.053 + 3 × 0.008 + 4 × 0.001) = 0.378; sequencing the entire genome of each patient would reveal ~10 times as many lethal recessives on average.
Carrier couples
Some pairs of patients identified themselves as couples. In this set, we found several “carrier couples” in which both partners were heterozygous carriers for the same condition. Table 3 lists disorders identified in carrier couples at elevated risk of passing the carried disorder to their children.
Comparison to traditional screening guidelines
Among carrier states we detected, 76.7% were for diseases not included in ACOG carrier screening guidelines, whereas 69.0% were for diseases not included in ACMG guidelines. The ACMG’s inclusion of spinal muscular atrophy accounted for most of the discrepancy.
We detected 433 individuals who would not have been identified as disease carriers in accordance with conventional ethnicity-based screening paradigms ( Table 4 ). For example, a non-Jewish carrier for familial dysautonomia would not be identified if following guidelines that recommend screening only for those with Jewish ancestry—26.3% of familial dysautonomia carriers in our dataset did not report Jewish ancestry. Rates of non-Jewish carriers are even higher, an average of 40.7%, for diseases recommended by the ACMG. Furthermore, in Table 5 , we show the 10 diseases with the highest carrier frequency for each population with more than 500 tested individuals, along with ACOG/ACMG screening recommendations. In general, many prevalent diseases in each population are not currently recommended for screening by either the ACOG or the ACMG.
Discussion
Data quality and limitations
This study represents the first large-scale analysis of patients undergoing carrier screening for an extensive list of recessive disorders in a clinical setting. As such, the data provide the first report of carrier frequencies for many rare disorders in multiple ethnic groups and are an important resource for guiding diagnosis, treatment, and prevention of Mendelian disease.
Because our assay was based on targeted genotyping, we were able to find only carriers of particular disease-causing mutations, not all carriers of each disease. (Patient reports explain that genotyping can only establish risk reduction, not risk elimination.) Consequently, the disease frequencies we report should be considered lower bounds, as there are other disease-causing alleles not surveyed here. Further examination of each individual disease on an allelic basis will help elucidate important data points such as overall carrier frequency, disease penetrance, and clinical sensitivity.
Although our targeted genotyping data is not comprehensive, it is a significant improvement on carrier rate estimation based on disease incidence rates. Such estimation suffers from statistical challenges such as underreporting of mild phenotypes and embryonic/child mortality affecting the assumptions of Hardy–Weinberg equilibrium.24,29,30 In contrast, our data come from directly assaying carrier genotypes rather than relying on mapping from phenotypic incidence to genotype.
Comparison to previously published carrier frequencies
Table 5 shows, for each ethnic group with n > 500 in our data set, the 10 most commonly detected diseases, along with our estimates and literature estimates for carrier frequency. For a handful of disease/ethnicity pairings, we were unable to find a previously published estimate. For most diseases, particularly in the well-studied Northwestern European and Ashkenazi Jewish populations, our estimates are similar to previously published estimates of carrier frequency. However, there are some notable outliers.
We observed significantly higher carrier frequencies than expected for severe diseases in several populations. In particular, we find a cystic fibrosis carrier frequency of 1 in 40 among south Asians (95% confidence interval 1 in 29–64), in marked contrast to the published 1 in 118 rate.21 This corroborates reports that cystic fibrosis is under-reported in the South Asian population.22 Another notable outlier in both Ashkenazi Jews and East Asians is carnitine palmitoyltransferase II deficiency, which appears as a notably frequent disease (1 in 43 in Ashkenazi Jews and 1 in 378 in East Asians), although the literature marks it as very rare.23 Finally, we detect Smith–Lemli–Opitz syndrome (SLOS) at a significantly higher than expected rate in all European populations, in Hispanics, and Middle Easterners (1 in 123 from literature vs. 1 in 68 here; 95% confidence interval 1 in 60–78). Prior work suggests that a higher carrier frequency for SLOS than that computed from its birth prevalence is reasonable, because mutations in DHCR7, the causative gene for SLOS, may cause significant fetal mortality.24
In the African-American population, we observe significantly reduced carrier frequencies relative to literature statistics for 7 of the top 10 diseases, other than sickle cell disease, α-1-antitrypsin deficiency, and cystic fibrosis. Comparison to prior data in this population is confounded by significantly varying levels of ethnic admixture in the African-American population; it is possible that our study population is genetically distinct at these loci relative to prior studies.16 Although we have not attempted to estimate admixture proportions in our study patients, it may be an interesting avenue for future research.
Similarly, in the East Asian population, we find much lower carrier frequencies for Pendred syndrome (1 in 252 vs. 1 in 51) and Pompe disease (1 in 366.5 vs. 1 in 112). We suspect two independent causes for the discrepancy. First, general-population screening data are not available for these diseases in this population; consequently, statistics are often derived from targeted study populations enriched in the mutations in question.25 Second, the East Asian population displays sufficient genetic diversity (e.g., population structure among Han Chinese, Koreans, and Japanese) to have confounded results in earlier genetic studies.26 Our self-reported ethnicity data do not have sufficient detail to identify this structure.
We note that direct comparison between our results and the published literature can be difficult due to previously mentioned statistical considerations and differing population substructures. As an example, carrier frequencies in Middle Eastern subpopulations reported in previous literature can vary widely and could also be substantially different from frequencies in Americans of Middle Eastern origin. Therefore, the references in Table 5 are not meant to present an exhaustive review but to provide context for comparison. The specific ethnic distributions merit further examination because robust data are unavailable for many diseases.
Comparison to ACOG/ACMG guidelines
ACOG and ACMG guidelines aim to identify couples with a minimum a priori carrier risk, based on ethnic background or general-population disease prevalence. The ACOG recommends at least a subset of patients be offered carrier screening for CF, hemoglobinopathies, Tay–Sachs disease, familial dysautonomia, and Canavan disease.1,2,6,7,8 ACMG recommendations add SMA, Fanconi anemia Group C, Niemann–Pick disease type A, Bloom syndrome, mucolipidosis IV, and Gaucher disease type 1.
For their intended purposes, the ACOG and ACMG guidelines serve well. However, our results demonstrate that most of the heterozygous states we identified fall outside these guidelines ( Tables 4 and 5 ). Because the current thought is that the average individual is heterozygous for at least five recessive diseases, this is not surprising.15 By comparison, screening most Caucasians only for CF, as is recommended by the ACOG, yields a 4% positive rate. Focusing on a smaller number of conditions and on targeted ethnic groups means that most individuals at elevated risk are not identified.
In particular, the data show that a number of severe Mendelian disorders are more prevalent than commonly understood ( Table 5 ), are often present outside their characteristic ethnic groups ( Table 4 ), and are not covered by current screening guidelines ( Table 5 ). For example, we find a SLOS carrier frequency of 1 in 68.2 in our overall population, almost twice the previously reported 1 in 123 carrier frequency.27 Neither the ACOG nor the ACMG currently recommend offering screening for this severe condition. In contrast, the ACMG does recommend screening for SMA, with a similar worldwide carrier frequency of 1 in 57 (our data) or 1 in 54 (literature).28 Indeed, in the Northwestern European population, we find indistinguishable carrier frequencies for SLOS and SMA: 1 in 50.3 and 1 in 49.9, respectively. Carnitine palmitoyltransferase II deficiency presents another illustration of a disease with a frequency worthy of consideration of wider screening. In our Ashkenazi Jewish population, the carrier rate was higher than several other conditions currently included in screening guidelines ( Table 5 ).
Ethnicity-based screening model vs. universal screening
Historically, screening has focused on a limited disorder list primarily determined by self-reported ethnic group. Others1,2,6,7,8,17 have noted that this model will soon be inadequate, given: (i) 2010 US Census data that demonstrate sharp increases in individuals reporting mixed racial ancestry, particularly among the younger population approaching reproductive age;18 (ii) limitations of, and patient preferences against, use of racial and ethnic categorization in medicine;19,20 (iii) the possibility of unknown or unreported ancestry, due to limited family history knowledge, adoption, or other factors; and (iv) the decreasing cost of pan-ethnic screening due to advances in genomics.11 Indeed, most participating clinics applied the same testing panel regardless of ethnicity, phasing out the ethnicity-based screening paradigm in favor of a universal screening model.
We have presented perhaps the most accurate measurements to date of carrier frequencies for hundreds of recessive Mendelian alleles using a large, ethnically diverse clinical sample. In contrast, prior data suffer from the statistical irregularities inherent in aggregating the results of multiple studies conducted under different protocols. Those irregularities result not from mistakes by the authors. Instead, the relative ordering of disease frequencies can only be determined with confidence when they are all assessed on the same dataset and under the same conditions. This, to our knowledge, has never been accomplished on the scale of this study, where we have applied multiplex carrier testing to sample a large population at a large number of genetic loci simultaneously.
Implications of widespread expanded carrier screening merit further study. These may include increased partner testing via screening or sequencing methods, higher demand for genetic counseling services and genetics education for nongenetics providers, cost-effectiveness, and patient interest and anxiety.
Our data demonstrate that current ethnicity-based approaches of genetic screening are not optimally aligned with the real distribution of carrier frequencies for severe genetic disease. In particular, we find that numerous conditions, not currently suggested for screening by the ACOG and the ACMG, are prevalent; and that other diseases currently recommended for screening only in certain populations, such as sickle cell disease and Canavan disease, are in fact widely distributed outside their “home” populations. We believe that these facts should be considered alongside the rapidly decreasing cost of multilocus genetic testing in the design of future carrier screening guidelines.
Disclosure
P.P., J.R.M., and J.L.J. are advisors to Counsyl. The remaining authors are employees at Counsyl.
References
American College of Obstetricians and Gynecologists & American College of Medical Genetics. Preconception and Prenatal Carrier Screening for Cystic Fibrosis. Clinical and Laboratory Guidelines. American College of Obstetricians and Gynecologists: Washington, DC, 2001.
American College of Obstetricians and Gynecologists. Committee opinion no. 325: update on carrier screening for cystic fibrosis. Obstet Gynecol 2005;106:1465–1468.
Prior TW ; Professional Practice and Guidelines Committee. Carrier screening for spinal muscular atrophy. Genet Med 2008;10:840–842.
Samons MS . Early Experiences of Prenatal Genetic Counselors With Spinal Muscular Atrophy Carrier Testing [Thesis]. Brandeis University: Waltham, MA, 2010.
Sims CA, Callum P, Ray M, Iger J, Falk RE . Genetic testing of sperm donors: survey of current practices. Fertil Steril 2010;94:126–129.
American College of Obstetricians and Gynecologists. Committee opinion no. 432: spinal muscular atrophy. Obstet Gynecol 2009;113:1194–1196.
American College of Obstetricians and Gynecologists. Practice bulletin no. 78: hemoglobinopathies in pregnancy. Obstet Gynecol 2007;109: 229–237.
American College of Obstetricians and Gynecologists. Committee opinion no. 442: preconception and prenatal carrier screening for genetic diseases in individuals of Eastern European Jewish descent. Obstet Gynecol 2009;14:950–953.
Gross SJ, Pletcher BA, Monaghan KG ; Professional Practice and Guidelines Committee. Carrier screening in individuals of Ashkenazi Jewish descent. Genet Med 2008;10:54–56.
Scott SA, Edelmann L, Liu L, Luo M, Desnick RJ, Kornreich R . Experience with carrier screening and prenatal diagnosis for 16 Ashkenazi Jewish genetic diseases. Hum Mutat 2010;31:1240–1250.
National Human Genome Research Institute. DNA Sequencing Costs: Data from the NHGRI Large-Scale Genome Sequencing Program. http://www.genome.gov/sequencingcosts. Accessed 2 September 2011.
Srinivasan BS, Evans EA, Flannick J, et al. A universal carrier test for the long tail of Mendelian disease. Reprod Biomed Online 2010;21:537–551.
Wei S, Quigg MH, Monaghan KG . Is cystic fibrosis carrier screening cost effective? Community Genet 2007;10:103–109.
Musci TJ, Caughey AB . Cost-effectiveness analysis of prenatal population-based fragile X carrier screening. Am J Obstet Gynecol 2005;192:1905–12; discussion 1912.
McConkey E . Human Genetics: The Molecular Revolution, 1st ed. Jones and Bartlett: Sudbury, MA, 1993.
Parra EJ, Marcini A, Akey J, et al. Estimating African American admixture proportions by use of population-specific alleles. Am J Hum Genet 1998;63:1839–1851.
Lee SS, Mountain J, Koenig B, et al. The ethics of characterizing difference: guiding principles on using racial categories in human genetics. Genome Biol 2008;9:404.
U.S. Census Bureau. Statistical Abstract of the United States: 2011, 130th ed. Washington, DC, 2010.
Condit C, Templeton A, Bates BR, Bevan JL, Harris TM . Attitudinal barriers to delivery of race-targeted pharmacogenomics among informed lay persons. Genet Med 2003;5:385–392.
De Marco M, Cykert S, Coad N, et al. Views on personalized medicine: do the attitudes of African American and white prescription drug consumers differ? Public Health Genomics 2010;13:276–283.
Rohlfs EM, Zhou Z, Heim RA, et al. Cystic fibrosis carrier testing in an ethnically diverse US population. Clin Chem 2011;57:841–848.
Mei-Zahav M, Durie P, Zielenski J, et al. The prevalence and clinical characteristics of cystic fibrosis in South Asian Canadian immigrants. Arch Dis Child 2005;90:675–679.
Taroni F, Verderio E, Dworzak F, Willems PJ, Cavadini P, DiDonato S . Identification of a common mutation in the carnitine palmitoyltransferase II gene in familial recurrent myoglobinuria patients. Nat Genet 1993;4: 314–320.
Nowaczyk MJ, Waye JS, Douketis JD . DHCR7 mutation carrier rates and prevalence of the RSH/Smith-Lemli-Opitz syndrome: where are the patients? Am J Med Genet A 2006;140:2057–2062.
Park HJ, Shaukat S, Liu XZ, et al. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet 2003;40:242–248.
Sawai H, Nishida N, Mbarek H, et al. No association for Chinese HBV-related hepatocellular carcinoma susceptibility SNP in other East Asian populations. BMC Med Genet 2012;13:47.
Kelley RI, Hennekam RC . The Smith-Lemli-Opitz syndrome. J Med Genet 2000;37:321–335.
Sugarman EA, Nagan N, Zhu H, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet 2012;20:27–32.
Lacassie Y, Storment JM, Lazarin GA . Serendipitous diagnosis of mild recessive multiple epiphyseal dysplasia through parental-targeted screening test. Am J Med Genet A 2011;155A:3136–3138.
Unger S, Bonafé L, Superti-Furga A . Multiple epiphyseal dysplasia: clinical and radiographic features, differential diagnosis and molecular basis. Best Pract Res Clin Rheumatol 2008;22:19–32.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Table S1.
(PDF 237 kb)
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Lazarin, G., Haque, I., Nazareth, S. et al. An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: results from an ethnically diverse clinical sample of 23,453 individuals. Genet Med 15, 178–186 (2013). https://doi.org/10.1038/gim.2012.114
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2012.114
Keywords
This article is cited by
-
Clinical application value of expanded carrier screening in the population of childbearing age
European Journal of Medical Research (2023)
-
Carrier burden of over 300 diseases in Han Chinese identified by expanded carrier testing of 300 couples using assisted reproductive technology
Journal of Assisted Reproduction and Genetics (2023)
-
Next generation sequencing is a highly reliable method to analyze exon 7 deletion of survival motor neuron 1 (SMN1) gene
Scientific Reports (2022)
-
Assessing clinical utility of preconception expanded carrier screening regarding residual risk for neurodevelopmental disorders
npj Genomic Medicine (2022)
-
Evaluating the model of offering expanded genetic carrier screening to high school students within the Sydney Jewish community
Journal of Community Genetics (2022)
