
Abstract
The urea cycle consists of six consecutive enzymatic reactions that convert waste nitrogen into urea. Deficiencies of any of these enzymes of the cycle result in urea cycle disorders (UCDs), a group of inborn errors of hepatic metabolism that often result in life-threatening hyperammonemia. Argininosuccinate lyase (ASL) catalyzes the fourth reaction in this cycle, resulting in the breakdown of argininosuccinic acid to arginine and fumarate. ASL deficiency (ASLD) is the second most common UCD, with a prevalence of ~1 in 70,000 live births. ASLD can manifest as either a severe neonatal-onset form with hyperammonemia within the first few days after birth or as a late-onset form with episodic hyperammonemia and/or long-term complications that include liver dysfunction, neurocognitive deficits, and hypertension. These long-term complications can occur in the absence of hyperammonemic episodes, implying that ASL has functions outside of its role in ureagenesis and the tissue-specific lack of ASL may be responsible for these manifestations. The biochemical diagnosis of ASLD is typically established with elevation of plasma citrulline together with elevated argininosuccinic acid in the plasma or urine. Molecular genetic testing of ASL and assay of ASL enzyme activity are helpful when the biochemical findings are equivocal. However, there is no correlation between the genotype or enzyme activity and clinical outcome. Treatment of acute metabolic decompensations with hyperammonemia involves discontinuing oral protein intake, supplementing oral intake with intravenous lipids and/or glucose, and use of intravenous arginine and nitrogen-scavenging therapy. Dietary restriction of protein and dietary supplementation with arginine are the mainstays in long-term management. Orthotopic liver transplantation (OLT) is best considered only in patients with recurrent hyperammonemia or metabolic decompensations resistant to conventional medical therapy.
Genet Med 2012:14(5):501–507
Similar content being viewed by others

Introduction
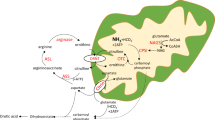
The urea cycle ( Figure 1 ) has two main functions: the detoxification of waste nitrogen into excretable urea and the de novo biosynthesis of arginine.1 Deficiencies of any of these enzymes of the cycle result in urea cycle disorders (UCDs), a group of inborn errors of hepatic metabolism that often result in life-threatening hyperammonemia. The overall prevalence of UCDs is estimated to be 1 in 8,200 in the United States.2

The urea cycle. The urea cycle consists of a series of steps catalyzed by enzymes (depicted in green boxes), which convert nitrogen from ammonia and aspartate into urea. ASLD leads to the accumulation of argininosuccinate upstream of the block as well as deficiency of arginine downstream of the block. ARG, arginase 1; ASL, argininosuccinate lyase; ASS, argininosuccinate synthase; CPS1, carbamoyl phosphate synthase 1; NAG, N-acetylglutamate; NAGS, N-acetylglutamate synthase; ORNT1, ornithine transporter; OTC, ornithine transcarbamylase. Reproduced with permission from publishers John Wiley and Sons.
The deficiency of the enzyme argininosuccinate lyase (ASL; MIM 608310), an enzyme that cleaves argininosuccinate to fumarate and arginine, results in an inborn error of ureagenesis termed argininosuccinic aciduria (OMIM 207900). ASL deficiency (ASLD) has an estimated prevalence of 1 in 70,000 live births,3 making it the second most common UCD.4 The first documented case of this condition was in 1958, when it was described as “a disease, probably hereditary, characterized by severe mental deficiency and a constant gross abnormality of amino acid metabolism.”5
Clinical Characteristics
The clinical presentation of ASLD is variable. The two most common forms are the severe neonatal-onset form and the late-onset form.
Severe neonatal-onset form
The clinical presentation of the severe neonatal-onset form is identical to that of other UCDs and is characterized by hyperammonemia within the first few days of life. Newborns typically appear healthy for the first 24 h but within the next few days develop vomiting, lethargy, hypothermia and refuse to accept feeds.3 Tachypnea and respiratory alkalosis are the early findings that suggest hyperammonemia. A failure to recognize and treat the defect in ureagenesis leads to worsening lethargy, seizures, coma, and death. The findings of hepatomegaly and trichorrhexis nodosa (coarse and friable hair) at this early stage are the only clinical findings that may suggest the diagnosis of ASLD.3
Late-onset form
The manifestations of the late-onset form range from episodic hyperammonemia (triggered by acute infection or stress or by noncompliance with dietary restrictions and/or medication) to cognitive impairment, behavioral abnormalities, and/or learning disabilities in the absence of any documented episodes of hyperammonemia.3
Long-term complications
Although manifestations secondary to hyperammonemia are common to all UCDs, many individuals with ASLD present with a more complex and unique clinical phenotype. There are increased incidences of (i) neurocognitive deficiencies, (ii) hepatic disease, and (iii) systemic hypertension in patients with ASLD. These manifestations appear to be unrelated to the severity or duration of the hyperammonemic episodes.6,7,8
Neurocognitive deficiencies
In a cross-sectional study of individuals with UCDs, it was observed that patients with ASLD had a significant increase in disabilities and neurologic abnormalities as compared with persons with ornithine transcarbamylase deficiency.4 Individuals with ASLD also had an increased incidence of attention deficit hyperactivity disorder, developmental disability (intellectual disability, behavioral abnormalities, and/or learning disability), and seizures compared to those diagnosed with other UCDs.4 In recent long-term outcome studies in individuals with ASLD, intellectual disability, developmental delay, seizures, and abnormal electroencephalograms were noted even in those without metabolic decompensations.8,9
However, it is to be noted that, although the neurocognitive deficits are more common in ASLD than in other UCDs, they are not universally present.
Hepatic disease
Liver disease in individuals with ASLD also appears to be independent of the defect in ureagenesis. Hepatic involvement ranges from hepatomegaly to elevations of liver enzymes to severe liver fibrosis.4,7,10,11 Liver involvement has been noted even in individuals treated with protein restriction and arginine supplementation who had not experienced significant hyperammonemia.7,9 At present, no biochemical or molecular features help predict liver dysfunction in patients with ASLD. Given the potential direct toxicity of argininosuccinic acid on hepatocytes, lowering the argininosuccinate levels in plasma (a reflection of its production by the liver) may have potential benefits, but this has not yet been proven.
Hypertension
Recently, it was noted that hypertension is overrepresented in persons with ASLD.12 Usually no secondary causes of hypertension are detected, suggesting that this finding is related to ASLD.
Electrolyte imbalances
Some individuals develop electrolyte imbalances such as hypokalemia (unpublished data). Hypokalemia is observed even in individuals who are not treated with sodium phenylbutyrate. The etiology is unclear; however, increased renal wasting has been suggested.
Other manifestations
Trichorrhexis nodosa is characterized by nodular swellings of the hair shaft accompanied by frayed fibers and loss of cuticle. About half of individuals with ASLD have an abnormality of the hair that manifests as dull, brittle hair surrounded by areas of partial alopecia13 as either an early or late manifestation. Normal hair contains 10.5% arginine by weight; hair that is deficient in arginine as a result of ASLD is weak and tends to break.
Biochemical Characteristics
Elevated plasma ammonia with respiratory alkalosis is the classical finding observed during periods of metabolic decompensations. Plasma amino acid analyses typically reveal elevated citrulline levels in the range of 100–300 μmol/l.3 The accumulation of argininosuccinic acid (and its anhydrides), the substrate upstream of the metabolic block is the biochemical hallmark of ASLD. The typical levels observed in ASLD patients range between 50 and 110 μmol/l in the plasma and >10,000 μmol/g of creatinine in the urine.8 Elevations of alanine, glutamine, and glycine, which imply deficiency of nitrogen disposal by conversion to urea, are also commonly observed in the plasma amino acid profile. Although typically the orotic acid excretion is in the normal ranges, orotic aciduria may be observed in ASLD.14,15 The impaired recycling of ornithine seems to contribute to the increase in carbamoyl phosphate that leads to overproduction of orotic acid.14
Molecular Characteristics
Normal allelic variants
The human ASL gene cloned in 1986 is located on chromosome 7q11.21.16 ASL consists of 17 exons with the first exon encoding for the 5′-untranslated region.17 A pseudogene Ψ ASL2 was recently mapped to a region ~3 Mb upstream of ASL. The pseudogene includes intron 2, exon 3, and part of intron 3 of ASL.17 The ASL gene has now been identified in a variety of species, including bacteria, Saccharomyces, algae, amphibia, rats, and humans.18 In humans, the protein is expressed predominantly in the liver but is also expressed in most other tissues, such as fibroblasts, kidney, heart, brain, muscle, pancreas, and red blood cells.
Normal gene product
ASL complementary DNA encodes a deduced protein of 464 amino acids with a predicted molecular mass of 52 kD. The active enzyme is a homotetramer of four identical subunits with a molecular mass of 208 kD, with four active sites in each tetramer.19 In addition to its role in ureagenesis, ASL is the only enzyme in the body that is capable of generating arginine. The expression pattern in a wide variety of tissues is likely intended to meet the need for arginine synthesis in these tissues.
ASL belongs to a superfamily of enzymes that have homologous motifs and catalyze similar cleavages with the release of fumarate as one of the products. Of all the enzymes in this family, ASL is most closely related to δ-crystallin, a protein found in abundance in the lens of birds and reptiles.20,21,22
Abnormal allelic variants
ASLD is caused by many heterogeneous mutations in the ASL gene. The enzymatic activity of ASL requires the assembly of four ASL monomers to form a homotetramer. Hence, the phenotypic consequences of a specific mutation are dependent on the mutation on the other allele and their ability to complement each other. The pathogenic mutations include nonsense and missense point mutations, insertions, deletions, and those affecting messenger RNA splicing. Mutations are scattered throughout the gene; however, exons 4, 5, and 7 appear to be mutational hotspots.17,23 Although most of the pathogenic alterations are “private mutations” in the families, there are three mutations with a founder effect.
-
The c.1153C>T variant, the founder mutation in the Finnish population, is associated with residual ASL enzyme activity as measured by the incorporation of [14C] citrulline into proteins.24
-
Two founder mutations are present in people of Arab ancestry from the Kingdom of Saudi Arabia: the c.1060C>T change that results in a premature stop codon is responsible for ~50% of the ASL mutations in this population;25 the other, c.346C>T, is common, but the proportion of individuals with this mutation is not known.
Abnormal gene product
Mutations in ASL lead to enzyme with reduced or no catalytic activity.
Molecular Pathogenesis
It is unlikely that elevated plasma ammonia is the only toxic compound in ASLD. The neurocognitive deficiencies can be observed even in the absence of hyperammonemia and liver dysfunction and hypertension occur seem unrelated to metabolic decompensations.8,9,12 These support the hypothesis that the phenotype in ASLD is likely due to a combination of the increase in argininosuccinic acid that is upstream of the block along with decreased endogenous synthesis of arginine that parsimoniously may lead to a decrease in arginine metabolites in various tissues.
Arginine is a semi-essential amino acid. The sources of arginine are exogenous from the diet and endogenous from the breakdown of proteins and synthesis from citrulline.26 In healthy adults, the level of endogenous synthesis (which is dependent on ASL) is sufficient to meet the arginine requirements of the body, thus making arginine a nonessential amino acid. However, in situations of catabolic stress or renal or intestinal dysfunction, endogenous arginine production is not commensurate with metabolic requirements and the body is dependent on exogenous sources.
The arginine generated in the liver, the major site of arginine metabolism, is converted to urea and ornithine. Thus, the liver does not contribute to the circulating pool of arginine. Approximately 60% of net synthesis of arginine in adult mammals occurs in the kidney, where citrulline is extracted from the blood and converted to arginine by the action of the enzymes ASS and ASL, located in the proximal tubules.27 However, many other tissues and cell types also contain both these enzymes for generating arginine from citrulline.28 In ASLD, arginine becomes an essential amino acid because all the cells and tissues are deficient in the ASL enzyme.
Arginine is the precursor for synthesis of many biologically important compounds including urea, nitric oxide (NO), polyamines, proline, glutamate, creatine, and agmatine ( Figure 2 ). With deficiency of ASL and the resulting deficiency of arginine, one could hypothesize that there would also be deficiency of NO and other metabolites for which it is a precursor. However, ASLD patients are supplemented with arginine and hence, theoretically, should not be deficient in its metabolites. The “arginine paradox” describes the observation that, despite apparently saturating intracellular levels of arginine, exogenously administered l-arginine is able to increase NO production. This important paradox suggests that l-arginine availability at the site of NO production may be the limiting factor.29,30 Thus, it has been hypothesized that compartmentalization and intracellular metabolite channeling underlie the arginine paradox that distinguishes the extracellular from the intracellular pools of arginine.

Metabolic fates of arginine. Arginine is derived from dietary sources, protein catabolism, and endogenous synthesis. The citrulline–NO cycle (green) is responsible for the regeneration of arginine in various tissues. Arginine serves as the precursor for many biologically important molecules; a decrease in arginine may result in decreased production of compounds for which it serves as a precursor. ADC, arginine decarboxylase; Arg1, arginase 1; ASA, argininosuccinic acid; ASL, argininosuccinate lyase; ASS, argininosuccinate synthase; GABA, γ-amino butyric acid; GATM, glycine amidinotransferase; NO, nitric oxide; NOS, NO synthase; OTC, ornithine transcarbamylase. Reproduced with permission from publishers John Wiley and Sons.
A hypomorphic mouse model of ASL deficiency demonstrates multiorgan dysfunction, including hypertension consistent with systemic NO deficiency.31 The mice have decreased NO production as evidenced by a significant decrease in S-nitrosylation and/or nitrite in heart and other tissues. Corroboratively, NO production in patients with ASLD is lower than in those with ornithine transcarbamylase or argininosuccinate synthase deficiency.31,32 Dynamic measurements of metabolite fluxes using stable isotopes in ASL-deficient patients as well as in an argininosuccinic acid mouse model reveal decreased NO flux.31 We have recently shown that there is a structural requirement of ASL for channeling of arginine into a protein complex necessary for NO production, and hence ASLD leads to a decrease in NO at the tissue and organism levels.31
Depletion of arginine as substrate for NO synthesis can cause increased free radical production due to uncoupling of NO synthase.33 An increase in free radical production can result in tissue damage, with the brain being particularly sensitive to both direct and indirect effects of free radicals mediated by increases in intracellular free Ca2+ and/or excitatory amino acids.34 Free radicals can interact with argininosuccinic acid in ASLD to form guanidinosuccinic acid, a cellular and neuronal toxin.35,36,37 It is yet to be determined if there is evidence for increased free radical production in ASLD.
Diagnosis
Newborn screening
All 50 states in the United States include ASLD in their newborn screening programs (2011 National Newborn Screening Status Report, http://genes-r-us.uthscsa.edu/nbsdisorders.pdf). Citrulline, assayed by tandem mass spectroscopy, is the metabolite used for detection of ASLD. Elevation of citrulline can also be seen with citrullinemia type 1 (ASS deficiency), citrullinemia type 2 (citrin deficiency), and pyruvate carboxylase deficiency; hence, confirmation of the diagnosis of ASLD rests on further biochemical, molecular, and/or enzymatic testing.
Biochemical testing
Confirmation of a clinical suspicion of ASLD or an abnormal newborn screen result is typically accomplished by plasma amino acid analyses that reveal elevated citrulline levels (in the range of 100–300 μmol/l) and the biochemical hallmark of increased argininosuccinic acid in the plasma and/or urine.
Molecular genetic testing
ASL is the only gene in which mutations are known to be associated with ASLD.
Sequence analysis. Sequence analysis of the coding region of ASL detects mutations in about 90% of the individuals with clinical and biochemical diagnosis of ASLD.
Deletion/duplication analysis. Although array comparative genomic hybridization–based assays have recently become available to detect deletions or duplications of ASL, no data are available on the frequency of these rearrangements in ASLD.
Targeted mutation analysis. The ASL c.1153C>T variant accounts for ~60% of mutations in the Finnish population and is offered on a clinical basis in Finland.38
Enzymatic activity testing
ASL enzyme activity can be measured in cell homogenates from a flash-frozen liver biopsy or, more conveniently, from skin fibroblasts or red blood cells by one of two methods:
-
Forward reaction: addition of the substrate argininosuccinate followed by fluorometric measurement of the product fumarate.24
-
Reverse reaction: addition of the substrates 14C fumarate and unlabeled arginine followed by the assay of the product 14C argininosuccinate.
Because the residual in vitro ASL enzyme activity as measured by these direct methods does not seem to correlate with the clinical severity, alternative indirect assays have been evaluated.8
The citrulline incorporation test is an indirect in vivo method of assessing the ASL activity in fibroblast cell cultures. Incorporation of14C from l-[ureido-14C]citrulline and 3H from L-[3,4,5-3H(N)]-leucine into acid-precipitable material has been shown in at least one longitudinal study to be more sensitive in detecting residual ASL enzyme activity than the direct ASL enzyme activity assay performed on cell lysates. Hence, this assay is reported to have better correlation with the phenotype.8 However, because the citrulline incorporation test involves a skin biopsy and the differences in results of this test compared with the results of the direct ASL enzyme assay were not significant, it is debatable as to whether this test is of value in determining the prognosis in a given individual with ASLD.
Prenatal diagnosis
If the mutations in the ASL gene are known, prenatal diagnosis can be performed by mutation analysis on either chorionic villous tissue or the amniocytes.39 Elevated levels of argininosuccinic acid in the amniotic fluid can also reliably detect affected fetuses.40,41,42 Analysis of the enzyme activity by either direct methods from chorionic villus tissue or amniocytes or indirect methods such as 14C-citrulline incorporation in uncultured chorionic villus samples have been successfully performed.24,43 Because of the limited amount of data available on the enzymatic analysis and argininosuccinic acid levels in the amniotic fluid, the sensitivity of these tests is not known. Moreover, the enzyme assays are available at only few specialized laboratories, precluding their use in clinical settings.
Genotype–phenotype correlation
The number of reported mutations is quite small compared with other urea cycle defects, as molecular genetic studies are not essential for the diagnosis.17,23 In general, there has been a poor correlation between the residual enzymatic activity and the severity of clinical phenotype. In vivo [14C] citrulline uptake has been reported to show better correlation with clinical phenotype in a single study, but this has not been yet replicated in larger cohort of patients.23 Recently, mutant ASL proteins were studied in an in vitro bacterial enzymatic assay,44 and the results were corroborated with data from clinical enzymatic testing. As observed with other methods, limited correlation was found between the patients’ clinical phenotype and the in vitro enzyme activity.
It is possible that the lack of correlation stems from a combination of decreased sensitivity of the current enzymatic assays and the fact that the different mutations may allow for complementation in the tetramer. In order to study these variables, different mutant alleles were characterized by evaluating the growth of yeast-deletion mutants in arginine-free media.45 This method can detect low levels of residual activity and assess the effect of different allelic complementation. The model was able to demonstrate that patients with late-onset form of ASLD either harbor significant residual activity or allow for intragenic complementation. In these late-onset ASLD patients, at least one active site was formed in the hybrid tetramer or the mutations partially stabilized each other.18,45 The drawback of the assay is its inability to ascertain the effect of the patient's genetic background on enzyme activity. Continued characterizations of different allelic combinations will allow further correlation between clinical phenotype and molecular changes.
Management
Evaluations following initial diagnosis
To establish the extent of the disease and the needs of an individual diagnosed with ASLD, the following evaluations are recommended: (i) complete neurocognitive evaluation; (ii) evaluation for evidence of hepatic involvement such as hepatomegaly, hepatitis, and signs of liver failure; and (iii) plotting of the systolic and diastolic blood pressure on the centile charts based on age and stature.
Treatment of manifestations
Treatment recommendations may be separated into two scenarios: (i) treatment for rapid control of hyperammonemia during metabolic decompensations and (ii) interval therapy to prevent the primary manifestations and long-term complications.
Treatment of acute metabolic decompensations
During acute hyperammonemic episodes severe enough to cause neurological symptoms, the treatment includes (i) discontinuing oral protein intake; (ii) supplementing oral intake with intravenous lipids, glucose, and intravenous insulin if needed (with close monitoring of blood glucose) to promote anabolism; and (iii) intravenous nitrogen-scavenging therapy.46
Intravenous nitrogen-scavenging therapy involves a loading dose of 600 mg/kg l-arginine-HCl and 250 mg/kg each of sodium benzoate and sodium phenylacetate in 25–35 ml/kg of 10% dextrose solution given intravenously over a 90-min period. This is followed by a sustained intravenous infusion of 600 mg/kg l-arginine-HCl and 250 mg/kg each of sodium benzoate and sodium phenylacetate over a 24-h period. When available, plasma concentrations of ammonia-scavenging drugs should be monitored to avoid toxicity. In the absence of drug levels, a serum anion gap of >15 mEq/l and an anion gap that has risen >6 mEq/l could indicate drug accumulation and increased risk for toxicity.
Ammonia levels usually normalize with therapy; however, failure to decrease ammonia levels with medical therapy mandates prompt institution of hemodialysis. Hemodialysis is the preferred method for rapid reduction of ammonia in patients who do not respond to nitrogen-scavenging therapy. Continuous arteriovenous hemodialysis or continuous venovenous hemodialysis with flow rates >40–60 ml/min is optimal. Some centers use extracorporeal membrane oxygenation with hemodialysis. Although this combination of techniques provides very high flow rates (170–200 ml/min) and rapidly reduces ammonia levels, morbidity is greater because of the need for surgical vascular access. Nitrogen-scavenging therapy needs to be continued during hemodialysis.
Prevention of primary manifestations
Dietary restriction of protein and dietary supplementation with arginine are the mainstays in the long-term management.
Diet. Lifelong dietary management is necessary and requires the services of a metabolic nutritionist. The recommended daily allowance for dietary protein is higher than the minimum needed for normal growth, and therefore most children with UCDs can receive less than the recommended daily allowance of protein and still maintain adequate growth. Plasma concentrations of ammonia, branched-chain amino acids, arginine plasma total protein, and serum albumin levels should be maintained within normal ranges. Plasma glutamine concentration should be maintained at <1,000 μmol/l if possible.3
Some of the correlations between compliance with the prescribed diet and outcome are contradictory. Although in some patients dietary therapy along with arginine supplementation have been shown to reverse the abnormalities of hair, improve cognitive outcome, and reverse electroencephalographic abnormalities,8,47,48 in many, dietary therapy has not been shown to influence the outcome of liver disease or cognitive impairment.7,9
Arginine base supplementation. The doses of arginine base routinely recommended are 400–700 mg/kg/day in persons weighing <20 kg and 8.8–15.4 g/m2 body surface area/day in those weighing >20 kg. Supplementation with arginine base helps replenish this amino acid, which is deficient in patients with ASLD, and promote excretion of nitrogen through the urea cycle as argininosuccinate. Arginine base is preferred for long-term chronic treatment because the long-term use of arginine hydrochloride may lead to hyperchloremic acidosis.
Arginine base supplementation has been shown to reverse the hair changes; however, its efficacy in preventing the chronic complications is not known. Although evidence suggests that arginine base supplementation may prevent metabolic decompensations in those with severe early-onset disease, long-term follow-up of persons identified through newborn screening programs did not detect a difference in outcomes between those who were supplemented with arginine base and those who were not.8,9,49 Because the renal clearance of argininosuccinic acid is high, increasing its production through arginine supplementation effectively increases waste nitrogen disposal, thereby decreasing the risk of hyperammonemia. However, given the theoretical risk of argininosuccinic acid toxicity on hepatocytes, reducing the amount of supplemental arginine by initiating nitrogen-scavenging therapy may have merits.
Oral nitrogen-scavenging therapy. Patients who have had metabolic decompensations or episodes of elevated ammonia despite being on a protein-restricted diet and arginine base supplementation are candidates for oral nitrogen-scavenging therapy, in which sodium benzoate and sodium phenyl butyrate stimulate the excretion of nitrogen in the form of hippuric acid and phenylacetylglutamine, respectively.49 The dose of sodium phenyl butyrate is 400–600 mg/kg/day for persons weighing up to 20 kg and 9–13 g/m2/day for those weighing >20 kg; the dose of sodium benzoate is 250–500 mg/kg/day.
Orthotopic liver transplantation
Long-term correction of ASLD in the liver can be accomplished by orthotopic liver transplantation (OLT),50 which has resulted in “biochemical cure.”51,52,53 However, OLT does not correct the arginine deficiency or the elevation of argininosuccinic acid in other tissues that are thought to account for the long-term complications of ASLD. Thus, we have recommended OLT only in patients with recurrent hyperammonemia or metabolic decompensations who are resistant to conventional medical therapy or in patients who develop cirrhosis with associated metabolic decompensations.
Prevention of secondary complications
Recommendations for salt restriction and use of antihypertensive therapy in those with elevated blood pressures are not different from those for the general population. Potassium supplementation must be considered in those with hypokalemia.
Surveillance
Regular monitoring of the concentration of plasma amino acids to identify deficiency of essential amino acids as well as impending hyperammonemia is essential. The appropriate intervals for monitoring depend on the clinical scenario, but they need to be more frequent in neonates and in those with frequent metabolic decompensations. We prefer to evaluate neonates every 1–2 weeks, infants who are between the ages of 2 months and 1 year every 1–3 months, and children aged >2 years every 3–4 months. Early signs of impending hyperammonemic episodes in older individuals include mood changes, headache, lethargy, nausea, vomiting, refusal to feed, ankle clonus, and elevated plasma concentrations of glutamine, alanine, and glycine. Plasma glutamine concentration may rise 48 h in advance of increases in plasma ammonia concentration in such individuals. Annual measurement of blood pressure using the appropriate-sized cuff and plotting of the centile values for age and stature would be important for diagnosing those with hypertension. Periodic evaluation of liver function tests and serum electrolytes may be necessary.
Agents/circumstances to avoid
The well-known precipitants for catabolism and/or hyperammonemia, including excess protein intake, prolonged fasting or starvation, and intravenous steroids, are to be avoided. Valproic acid can precipitate liver failure and is best avoided in these patients.
Genetic Counseling
ASLD is inherited in an autosomal recessive manner. The parents of an affected individual are obligate heterozygotes and are asymptomatic. At conception, each sib of an affected individual has a 25% chance of being affected. Although the phenotypic manifestations can be variable, in families with one child with the severe neonatal-onset form, subsequent children are likely to have the same form. By contrast, the phenotype of late-onset forms associated with partial ASL enzyme activity is variable.
Carrier detection
Carrier testing for at-risk family members is possible if the disease-causing mutations in the family have been identified. Carrier testing is not available by biochemical methods.
Disclosure
The authors declare no conflict of interest.
REFERENCES
Brusilow SW, Horwich AL . The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill: New York, 2001.
Brusilow SW, Maestri NE . Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr 1996;43:127–170.
Brusilow SW, Horwich AL . Online Metabolic and Molecular Basis of Inherited Disease. McGraw Hill Medical, (www.ommbid.com), 2009.
Tuchman M, Lee B, Lichter-Konecki U, et al. ; Urea Cycle Disorders Consortium of the Rare Diseases Clinical Research Network. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab 2008;94:397–402.
Allan JD, Cusworth DC, Dent CE, Wilson VK . A disease, probably hereditary characterised by severe mental deficiency and a constant gross abnormality of aminoacid metabolism. Lancet 1958;1:182–187.
Saudubray JM, Touati G, Delonlay P, et al. Liver transplantation in urea cycle disorders. Eur J Pediatr 1999;158(suppl 2):S55–S59.
Mori T, Nagai K, Mori M, et al. Progressive liver fibrosis in late-onset argininosuccinate lyase deficiency. Pediatr Dev Pathol 2002;5:597–601.
Ficicioglu C, Mandell R, Shih VE . Argininosuccinate lyase deficiency: longterm outcome of 13 patients detected by newborn screening. Mol Genet Metab 2009;98:273–277.
Mercimek-Mahmutoglu S, Moeslinger D, Häberle J, et al. Long-term outcome of patients with argininosuccinate lyase deficiency diagnosed by newborn screening in Austria. Mol Genet Metab 2010;100:24–28.
Billmeier GJ Jr, Molinary SV, Wilroy RS Jr, Duenas DA, Brannon ME . Argininosuccinic aciduria: investigation of an affected family. J Pediatr 1974;84:85–89.
Zimmermann A, Bachmann C, Baumgartner R . Severe liver fibrosis in argininosuccinic aciduria. Arch Pathol Lab Med 1986;110:136–140.
Brunetti-Pierri N, Erez A, Shchelochkov O, Craigen W, Lee B . Systemic hypertension in two patients with ASL deficiency: a result of nitric oxide deficiency. Mol Genet Metab 2009;98:195–197.
Fichtel JC, Richards JA, Davis LS . Trichorrhexis nodosa secondary to argininosuccinicaciduria. Pediatr Dermatol 2007;24:25–27.
Brosnan ME, Brosnan JT . Orotic acid excretion and arginine metabolism. J Nutr 2007;137(6 suppl 2):1656.S–1661S.
Gerrits GP, Gabreëls FJ, Monnens LA, et al. Argininosuccinic aciduria: clinical and biochemical findings in three children with the late onset form, with special emphasis on cerebrospinal fluid findings of amino acids and pyrimidines. Neuropediatrics 1993;24:15–18.
O’Brien WE, McInnes R, Kalumuck K, Adcock M . Cloning and sequence analysis of cDNA for human argininosuccinate lyase. Proc Natl Acad Sci USA 1986;83:7211–7215.
Trevisson E, Salviati L, Baldoin MC, et al. Argininosuccinate lyase deficiency: mutational spectrum in Italian patients and identification of a novel ASL pseudogene. Hum Mutat 2007;28:694–702.
Yu B, Howell PL . Intragenic complementation and the structure and function of argininosuccinate lyase. Cell Mol Life Sci 2000;57:1637–1651.
Turner MA, Simpson A, McInnes RR, Howell PL . Human argininosuccinate lyase: a structural basis for intragenic complementation. Proc Natl Acad Sci USA 1997;94:9063–9068.
Vallée F, Turner MA, Lindley PL, Howell PL . Crystal structure of an inactive duck delta II crystallin mutant with bound argininosuccinate. Biochemistry 1999;38:2425–2434.
Wistow G, Piatigorsky J . Recruitment of enzymes as lens structural proteins. Science 1987;236:1554–1556.
Yeh LS, Elzanowski A, Hunt LT, Barker WC . Homology of delta crystallin and argininosuccinate lyase. Comp Biochem Physiol, B 1988;89:433–437.
Linnebank M, Tschiedel E, Häberle J, et al. Argininosuccinate lyase (ASL) deficiency: mutation analysis in 27 patients and a completed structure of the human ASL gene. Hum Genet 2002;111:350–359.
Kleijer WJ, Garritsen VH, Linnebank M, et al. Clinical, enzymatic, and molecular genetic characterization of a biochemical variant type of argininosuccinic aciduria: prenatal and postnatal diagnosis in five unrelated families. J Inherit Metab Dis 2002;25:399–410.
Al-Sayed M, Alahmed S, Alsmadi O, et al. Identification of a common novel mutation in Saudi patients with argininosuccinic aciduria. J Inherit Metab Dis 2005;28:877–883.
Wu G, Morris SM Jr . Arginine metabolism: nitric oxide and beyond. Biochem J 1998;336 (Pt 1):1–17.
Windmueller HG, Spaeth AE . Source and fate of circulating citrulline. Am J Physiol 1981;241:E473–E480.
Mori M, Gotoh T . Arginine metabolic enzymes, nitric oxide and infection. J Nutr 2004;134(10 suppl):2820S–2825S; discussion 2853S.
Cui R, Iso H, Pi J, et al. Metabolic syndrome and urinary cGMP excretion in general population. Atherosclerosis 2007;190(2):423–428.
Vukosavljevic N, Jaron D, Barbee KA, Buerk DG . Quantifying the L-arginine paradox in vivo. Microvasc Res 2006;71:48–54.
Erez A, Nagamani S, Shchelochkov OA, et al. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nature Medicine 2011; e-pub ahead of print 13 November 2011.
Nagasaka H, Tsukahara H, Yorifuji T, et al. Evaluation of endogenous nitric oxide synthesis in congenital urea cycle enzyme defects. Metabolism 2009;58:278–282.
Pignitter M, Gorren AC, Nedeianu S, Schmidt K, Mayer B . Inefficient spin trapping of superoxide in the presence of nitric-oxide: implications for studies on nitric-oxide synthase uncoupling. Free Radic Biol Med 2006;41:455–463.
Halliwell B . Reactive oxygen species and the central nervous system. J Neurochem 1992;59:1609–1623.
Aoyagi K . Inhibition of arginine synthesis by urea: a mechanism for arginine deficiency in renal failure which leads to increased hydroxyl radical generation. Mol Cell Biochem 2003;244:11–15.
Aoyagi K, Shahrzad S, Iida S, et al. Role of nitric oxide in the synthesis of guanidinosuccinic acid, an activator of the N-methyl-D-aspartate receptor. Kidney Int (suppl) 2001;78:S93–S96.
D’Hooge R, Pei YQ, Marescau B, De Deyn PP . Convulsive action and toxicity of uremic guanidino compounds: behavioral assessment and relation to brain concentration in adult mice. J Neurol Sci 1992;112:96–105.
Keskinen P, Siitonen A, Salo M . Hereditary urea cycle diseases in Finland. Acta Paediatr 2008;97:1412–1419.
Häberle J, Koch HG . Genetic approach to prenatal diagnosis in urea cycle defects. Prenat Diagn 2004;24:378–383.
Kleijer WJ, Garritsen VH, van der Sterre ML, Berning C, Häberle J, Huijmans JG . Prenatal diagnosis of citrullinemia and argininosuccinic aciduria: evidence for a transmission ratio distortion in citrullinemia. Prenat Diagn 2006;26:242–247.
Kamoun P, Fensom AH, Shin YS, et al. Prenatal diagnosis of the urea cycle diseases: a survey of the European cases. Am J Med Genet 1995;55:247–250.
Mandell R, Packman S, Laframboise R, et al. Use of amniotic fluid amino acids in prenatal testing for argininosuccinic aciduria and citrullinaemia. Prenat Diagn 1996;16:419–424.
Pijpers L, Kleijer WJ, Reuss A, et al. Transabdominal chorionic villus sampling in a multiple pregnancy at risk of argininosuccinic aciduria: a case report. Am J Med Genet 1990;36:449–450.
Engel K, Vuissoz JM, Eggimann S, et al. Bacterial expression of mutant argininosuccinate lyase reveals imperfect correlation of in-vitro enzyme activity with clinical phenotype in argininosuccinic aciduria. J Inherit Metab Dis 2011. e-pub ahead of print 11 June 2011.
Trevisson E, Burlina A, Doimo M, et al. Functional complementation in yeast allows molecular characterization of missense argininosuccinate lyase mutations. J Biol Chem 2009;284:28926–28934.
Consensus statement from a conference for the management of patients with urea cycle disorders. J Pediatr 2001;138(1 suppl):S1–S5.
Kvedar JC, Baden HP, Baden LA, Shih VE, Kolodny EH . Dietary management reverses grooving and abnormal polarization of hair shafts in argininosuccinase deficiency. Am J Med Genet 1991;40:211–213.
Coryell ME, Hall WK, Thevaos TG, et al. A familial study of a human enzyme defect, argininosuccinic aciduria. Biochem Biophys Res Commun 1964;14:307–312.
Batshaw ML, MacArthur RB, Tuchman M . Alternative pathway therapy for urea cycle disorders: twenty years later. J Pediatr 2001;138(1 suppl):S46–54; discussion S54.
Lee B, Goss J . Long-term correction of urea cycle disorders. J Pediatr 2001;138(1 suppl):S62–S71.
Robberecht E, Maesen S, Jonckheere A, Van Biervliet S, Carton D . Successful liver transplantation for argininosuccinate lyase deficiency (ASLD). J Inherit Metab Dis 2006;29:184–185.
Marble M, McGoey RR, Mannick E, et al. Living related liver transplant in a patient with argininosuccinic aciduria and cirrhosis: metabolic follow-up. J Pediatr Gastroenterol Nutr 2008;46:453–456.
Newnham T, Hardikar W, Allen K, et al. Liver transplantation for argininosuccinic aciduria: clinical, biochemical, and metabolic outcome. Liver Transpl 2008;14:41–45.
Acknowledgements
This work was supported by the NIH (DK54450, RR19453, RR00188, GM90310 to B.L., GM07526, and DK081735 to A.E.). A.E. was supported by the NUCDF fellowship. S.C.S.N. is supported by fellowship grants from the NUCDF and the LCRC of the Osteogenesis Imperfecta Foundation. We acknowledge and are grateful for the clinical efforts of Mary Mullins, Susan Carter, Alyssa Tran, Janice Stuff, and the TCH General Clinical Research Center nursing staff.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nagamani, S., Erez, A. & Lee, B. Argininosuccinate lyase deficiency. Genet Med 14, 501–507 (2012). https://doi.org/10.1038/gim.2011.1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2011.1
Keywords
This article is cited by
-
Cardiac disruption of SDHAF4-mediated mitochondrial complex II assembly promotes dilated cardiomyopathy
Nature Communications (2022)
-
ASS1 and ASL suppress growth in clear cell renal cell carcinoma via altered nitrogen metabolism
Cancer & Metabolism (2021)
-
Establishment of the circadian metabolic phenotype strategy in spontaneously hypertensive rats: a dynamic metabolomics study
Journal of Translational Medicine (2020)
-
Free Radical Scavengers Prevent Argininosuccinic Acid-Induced Oxidative Stress in the Brain of Developing Rats: a New Adjuvant Therapy for Argininosuccinate Lyase Deficiency?
Molecular Neurobiology (2020)
-
Low prevalence of argininosuccinate lyase deficiency among inherited urea cycle disorders in Korea
Journal of Human Genetics (2018)
