
Abstract
ACMG previously developed recommendations for standards for interpretation of sequence variations. We now present the updated revised recommendations. Here, we describe six interpretative categories of sequence variations: (1) sequence variation is previously reported and is a recognized cause of the disorder; (2) sequence variation is previously unreported and is of the type which is expected to cause the disorder; (3) sequence variation is previously unreported and is of the type which may or may not be causative of the disorder; (4) sequence variation is previously unreported and is probably not causative of disease; (5) sequence variation is previously reported and is a recognized neutral variant; and (6) sequence variation is previously not known or expected to be causative of disease, but is found to be associated with a clinical presentation. We emphasize the importance of appropriate reporting of sequence variations using standardized terminology and established databases, and of clearly reporting the limitations of sequence-based testing. We discuss follow-up studies that may be used to ascertain the clinical significance of sequence variations, including the use of additional tools (such as predictive software programs) that may be useful in variant classification. As more information becomes available allowing the interpretation of a new sequence variant, it is recommended that the laboratory amend previous reports and provide updated results to the physician. The ACMG strongly recommends that the clinical and technical validation of sequence variation detection be performed in a CLIA-approved laboratory and interpreted by a board-certified clinical molecular geneticist or equivalent.
Similar content being viewed by others

Main
These recommendations for the standardization of interpretation and reporting of sequence variations identified in the course of providing clinical laboratory services are intended (1) to provide a framework for laboratories for the interpretation and reporting of such test results, and (2) to aid referring clinicians by educating them as to possible testing outcomes so that they may inform their patients and families appropriately. These revised recommendations are based on the foundation laid by the previous ACMG practice guideline recommendations in 2000.1
I. INTERPRETATIVE CATEGORIES AND DEFINITIONS OF SEQUENCE VARIATIONS
Increasingly, clinical molecular laboratories are detecting novel sequence variations in the course of testing patient specimens. The certainty with which any given sequence variation is of clinical significance falls within a spectrum of interpretations, ranging from those in which the variation is almost certainly of clinical significance to those in which it is almost certainly not. The ACMG Laboratory Quality Assurance Committee Working Group on Standards for Interpretation of Sequence Variations Revisions 2007 recommends the following interpretative categories of sequence variations for the purposes of clinical reporting.
1. Sequence variation is previously reported and is a recognized cause of the disorder
Review of the literature, central mutation databases, e.g., Human Gene Mutation Database,2 or appropriate locus-specific databases, to assess the current degree of certainty that the sequence variation is causative of the disorder should be undertaken before reporting.
2. Sequence variation is previously unreported and is of the type which is expected to cause the disorder
Examples include variation that is predicted to result in the introduction of a stop codon or a missense mutation of the normal stop codon; mutate the initiation codon (ATG); alter the sequence at a splice junction, particularly the invariant AG/GT nucleotides; or delete one or more nucleotides or exons in such a manner as to lead to a shift in the mRNA reading frame.
3. Sequence variation is previously unreported and is of the type which may or may not be causative of the disorder
Examples include a variation that is located within a splice consensus sequence, likely to produce a cryptic splice site, likely to affect transcription, any missense mutation, in-frame amino acid insertion/deletion, or in-frame exon deletion. A missense mutation that leads to a nonconservative substitution of an evolutionarily conserved amino acid is more likely to be causative of the disorder than a missense mutation that leads to a conservative substitution or alters an amino acid that is not evolutionarily conserved. Family studies and structural analysis of the protein may clarify the pathologic or benign nature of missense mutations (see III, 1 below).
4. Sequence variation is previously unreported and is probably not causative of disease
Examples include sequence variations that do not produce an amino acid substitution and that are unlikely to produce a cryptic splice site, sequence changes that occur in the intron, far from the intron/exon boundary, and are unlikely to affect splicing, sequence changes seen in conjunction with a second deleterious sequence change, such as a nonsense or a frameshift mutation (in the case of dominant disorders), sequence changes seen multiple times in different samples through clinical testing or seen in an unaffected parent.
5. Sequence variation is previously reported and is a recognized neutral variant
Review of the literature, central mutation databases, e.g., Human Gene Mutation Database,2 dbSNP,3 and appropriate locus-specific databases, to assess the current degree of certainty that the sequence variation is a recognized neutral variant should be undertaken before reporting.
6. Sequence variation is not known or expected to be causative of disease, but is found to be associated with a clinical presentation
If there is significant evidence in the literature, functional evidence, or multiple published reports, these sequence variations should be reported with a brief explanation of their relevance to the disorder. An example in this category would be rare alleles coding for a silent polymorphism (Ala45Ala, Leu769Leu), which have been found to be associated with Hirschsprung Disease4 in the absence of a disease-associated mutation. The rare alleles at this position may predispose to Hirschsprung Disease in a complex, low-penetrance fashion, and may modify expression of the disease phenotype. It should be made clear that these are not definitive mutations, that the evidence comes from population studies, and that medical management and family planning decisions should not be made based on them.
The extent to which a sequence variation is considered causative of disease may be influenced by a number of other parameters. A partial list includes clinical presentation, the individual's risk of having the disorder, family history, segregation of the variant with affectedness in a family, nature, and position of the amino acid substitution, evolutionary conservation of the affected residue, co-occurrence with a deleterious mutation, epidemiological and case/control studies, evaluation of chromosomes from equivalent population, functional in vitro studies, knock-out or knock-in animal models, other test results, and the sensitivity and specificity of the test being performed. All of these studies must be interpreted cautiously.
II. REPORTING OF SEQUENCE VARIATIONS
The purpose of test reports is to state clearly the presence, if detected, of any sequence change that differs from the consensus wild-type sequence and may be of clinical significance. Common, well-known polymorphisms detected often in a particular gene, with which the testing laboratory is familiar and which has previously been established as benign, need not be reported. Reports should indicate that common polymorphisms have not been included in the report. Variants of unknown clinical significance, whether believed to be benign or not, must be included in the report and followed by the laboratory's interpretation of the likely clinical significance. Both the ACMG Standards and Guidelines for clinical genetics laboratories5 and the CAP Molecular Pathology Checklist6 explicitly state that the laboratory must provide the interpretive information and a best estimate of clinical significance for the variant(s) as part of the report. Reports should be written in a style that is clear and informative to the clinician and does not require in-depth knowledge of molecular genetics to be interpretable and useful. Guidelines for report content are presented below.
-
1
Reports should identify the gene analyzed, indicate the absence or presence of a sequence variation other than those judged to be of no consequence, and, if applicable, indicate the location of the sequence variation by nucleotide position, codons affected, deduced amino acid change(s), and whether the substitution is conservative or nonconservative from a biochemical perspective. They should also include whether all exons or only targeted exons were sequenced and whether intron-exon boundaries were analyzed.
-
2
Reports should indicate the interpretative category of the test results (see above I, 1–6). Each laboratory needs to determine the level of comfort and the granularity of these categories for each gene it analyzes. The category of novel missense variant of uncertain significance may be further subdivided into “suspected deleterious change” and “suspected benign variant” if enough supportive data exist to allow for these nuances. Because this information may be used for medical decisions, such as surgery or pregnancy termination, the recommended conservative approach is to avoid speculation and simply classify the variant as one of unknown clinical significance, even if potentially more frustrating for the patient.
-
3
Reports should provide a discussion of the basis upon which the interpretation was made with citation of available supportive data (see above).
-
4
Reports should indicate the methodology and its analytical sensitivity used in the analysis in sufficient detail to permit the clinician to determine the analytic reliability of the observation (e.g., detection by a mutation scanning technique, such as denaturing high-pressure liquid chromatography or high-resolution melting alone, or in combination with targeted sequence confirmation, or by direct sequence analysis of the coding region of the gene and flanking intron/exon boundaries). Also see section on “limitations of mutation-detection testing ” (Section B).
-
5
Reports in which a previously reported sequence variant was identified should include information on penetrance, expressivity, etc., or state that no relevant data exist.
-
6
Reports in which a sequence variant not previously reported was identified, and for which no relevant data exist, should include possible strategies for studying the variant further in an effort to arrive at a clinical interpretation. At the very least, this should include a recommendation to test other affected and unaffected family members to determine whether the variant segregates with the phenotype within the family. Further extensive work would include application of algorithms (Fig. 1).
Fig 1 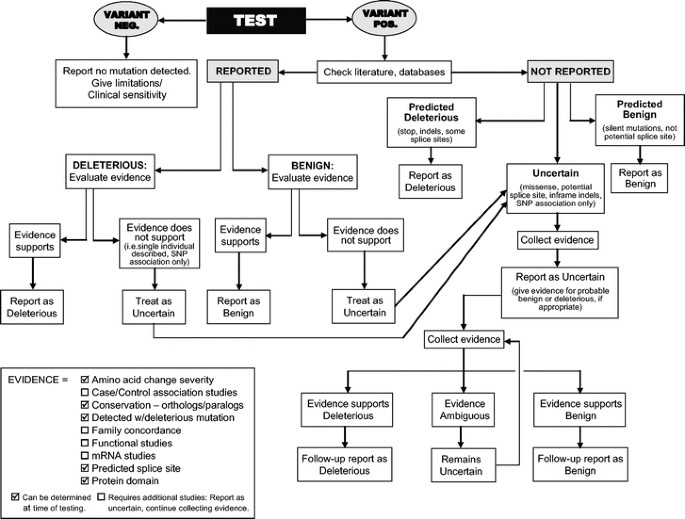
A decision tree for interpretation of sequence variants and clinical reporting. Evidence that can be used to support sequence variant interpretation is shown in the box at the bottom.

A. Use of standardized terminology and established databases for reporting sequence variants
1. Nomenclature
A uniform gene mutation nomenclature is recommended for accurate reporting of mutations in clinical reports. Standard gene mutation nomenclature7 has been described. Not all types of mutations (e.g., complex mutations) are covered by these recommendations, but possible descriptions for complex mutations have been reported.8 Clinical reports should describe the level at which the mutation is being described, e.g., “g” for genomic sequence, “c” for complementary DNA sequence, “p” for protein, etc.9 (http://www.hgvs.org/mutnomen/).
2. Reference sequence
A standard reference sequence for each gene should be used for describing mutations and noted in the report. The reference sequence should be complete and derived from the NCBI refseq database10 and the version number. If a genomic sequence is used, it should cover the entire gene (including the 5′ and 3′ UTR sequences). For mutations described using coding DNA reference sequence the appropriate transcript should be used and the mutation should be described from the nucleotide 1 “A” of the ATG-translation initiation codon. The origin of the transcript used should be specified, e.g., the most common human transcript, largest known transcript, or tissue-specific alternatively spliced transcript.
3. Databases
A large number of databases are now available, which curate the growing number of disease mutations and useful polymorphisms being discovered in the human genome. In determining and reporting the classification of a variant, clinical laboratories may find valuable information in databases as well as in published literature. Sequence databases are useful in finding reference sequences and identifying reported variants. Disease-specific databases often list reported variants along with the published references. However, these databases were not initially designed to meet clinical standards and should be used with caution. Curators have found significant literature errors when curating gene- or locus-specific databases. In using these databases, clinical laboratories should note the following:
-
1
Any nonstandard numbering systems, numbering start sites, or variant designations;
-
2
The frequency with which the database is curated and the latest update;
-
3
Evidence for variant classification (it is recommended to check the original cited reference);
-
4
Number of times the variant has been reported;
-
5
Populations in which a variant has been reported.
Once an assay has been transferred from research into the clinical setting, the clinical laboratories become a rich source for new variant discovery. The goal of the Human Variome Project11 is the worldwide collection and classification of all gene variants, whether pathologic or benign. Clinical laboratories are encouraged to contribute to gene mutational databases. Clinical information should be provided whenever possible, following Health Insurance Portability and Accountability Act12 regulations for privacy. Evidence of variant classification should also be submitted. We recognize, however, that clinical laboratories may not easily implement collecting of evidence and submitting them to databases. Clinical laboratories are encouraged to form collaborations with clinicians to provide clinical information for gene-specific databases to allow a better understanding of how genotype influences clinical phenotype. Because of the great potential of these databases to aid clinical laboratory practice, efforts are underway for clinical mutational databases to be developed and standardized. Standardization should provide easier access to updated information as well as easier submissions for the clinical laboratory. The clinical laboratory should have an internal system to track all sequence variations identified in each gene. This is important for tracking genotype/phenotype correlations and the frequency of variants in affected and normal populations. If the interpretation must be modified or updated for a novel sequence variant or variation previously thought to be benign (see Section IV), the laboratory will need to identify all patients with the particular sequence variant. It is also useful to track polymorphisms associated with mutations as these could possibly modify the phenotype, help identify founder effects, or be used for linkage analysis. It is not unusual to find a new sequence variation in an affected individual that turns out to be an ethnic-specific polymorphism. Additional information useful to include in internal databases is exon or intron number, clinical phenotype, and ethnicity.
B. Limitations of sequence-based testing should be reported
-
1
Normal sequence variation in the genome can affect results obtained by sequence-based testing and should be taken into account when reporting sequence-based test results. For example, the presence of a normal sequence variation (polymorphism) within the polymerase chain reaction (PCR) primer binding site may affect amplification and subsequently the sequencing result. If a mutation occurs on the allele with the (primer-binding site) polymorphism, it may not be detected if that allele fails to amplify. Although appropriate care should be taken to design primers in nonpolymorphic regions of the genome, this is not always possible because many polymorphisms are not reported and indeed may not yet have been identified. It is therefore important to indicate while reporting sequencing results (especially for normal results or when only one mutation is found when two are expected as in autosomal recessive diseases) that polymorphic/normal genomic variation in the patient sample may interfere with mutation detection.
-
2
Large gene deletions that involve one or several exons will not be detected by sequence analysis as the primer-binding sites for the amplification primers are deleted. The presence of a large deletion when present on one allele will be masked by the amplified normal allele. It is therefore important to indicate while reporting results (especially for normal results or when only one mutation is found when two are expected as in autosomal recessive diseases) that certain types of mutations, such as large deletions will be missed by sequencing (or other PCR-based mutation scanning method used).
-
3
The presence of a rare polymorphism or mutation in the apparent homozygous state may be indicative of nonamplification of one allele (because of either a polymorphism in the primer binding site or a deletion of that allele) and may, in actuality, represent hemizygosity for the polymorphism. In the case of an autosomal recessive disorder, the presence of homozygosity for a rare mutation could be attributed to the presence of consanguinity in the family or a founder effect. Follow-up studies using different primers for PCR/sequencing or deletion analysis or parental DNA (with appropriate consent) should be considered.
-
4
The sensitivity of the mutation detection technology used to detect sequence changes should also be mentioned in the patient report. For example, sequencing has an analytical sensitivity of approximately 99% for the detection of nucleotide base alterations, small deletions, and insertions, but it does not detect large deletions, thus affecting the clinical sensitivity for the disease. Low-level mosaicism will not be detected by routine sequencing methodologies.
-
5
The report should also indicate the regions of the gene covered by the mutation detection technique used and the fact that mutations that occur outside of these regions would not be detected. For example, if only the coding and immediate flanking regions of genes are analyzed, new splice sites, changes in the promoter region, and other noncoding or regulatory regions will not be detected.
III. FOLLOW-UP STUDIES TO ASCERTAIN THE CLINICAL SIGNIFICANCE OF SEQUENCE VARIATIONS
Classification of sequence variants into high-risk or low-risk categories is both challenging and critical for clarification of causative status. When the causation status of a sequence variant is indeterminate, follow-up activities may be useful to clarify this relationship and assist with risk assessment.
-
1
Assessment of the likely clinical significance may be improved by testing additional family members. Relationships, e.g., parentage, sibships, and more distantly related family members should be confirmed as appropriate. Family concordance studies are useful for clarifying the relationship between sequence variation and phenotype in all Mendelian disorders, no matter what the inheritance pattern. The following paragraph deals with the case of a new mutation in the proband.
-
2
In cases in which the phenotype is de novo within the family, parental DNA can be evaluated for the presence or absence of the sequence variation identified previously in the proband. For X-linked recessive and autosomal dominant traits, absence of the sequence variation in either parent significantly increases the likelihood that the sequence variation is causative of the proband's phenotype. Conversely, the presence of the sequence variation in both an unaffected parent and the proband is of uncertain interpretation. Consideration of age of onset and phenotypic variation should be appropriately evaluated in this assessment.
-
3
Sequence variations, independent of clarification of their causation status, may be used in classical linkage analysis for carrier, predictive, and prenatal testing. In cases in which there is a family history of the disorder, affected and unaffected family members can be evaluated for the presence of the sequence variation and the results subjected to linkage analysis. The accuracy of the analysis is sensitive to those sources of error inherent in linkage analyses, e.g., undetected recombination, locus heterogeneity, and availability of appropriate family members for testing.
-
4
In recessive disorders, inheritance of two sequence variants in trans should be confirmed by analysis of parental DNA if possible. Association of two variants in cis removes the possibility that they are by themselves a complete explanation of an autosomal recessive phenotype.
-
5
Extensive testing of normal chromosomes or previous experience of the laboratory in not finding a novel variant in a normal population may be helpful in determining whether the variant is disease-causing or benign.
Additional tools that can be used to assist in variant classification include the following:
-
1
Indirect measures such as Grantham analysis13 of the fit between the variant and the evolutionary range of variation at that position, estimation of splice site robustness derived from other model systems, and the location and perceived severity of amino acid change as related to functional domains. Software programs such as SIFT,14 Polyphen,15 and Conseq16 can be used to help predict the effect of amino acid changes. Multiple software predictive programs are available to predict splicing (creation or loss of site and intronic or exonic) such as Splice Site Prediction by Neural Network17 (http://www.fruitfly.org/seq_tools/splice.html) or ESEfinder:Exonic Splicing Enhancer Finder18 (http://rulai.cshl.edu/tools/ESE/index.html). Laboratories should validate any predictive software program used clinically.
-
2
Evidence from functional analysis such as pathologic classification, histopathology and immunochemical profiles, protein function assays, structural analysis (modeling), loss of heterozygosity and analysis of RNA species when determination that the aberrant product is derived from the affected allele is possible.
Each of these tools can be used as an independent estimator of risk. Additionally, multifactorial likelihood models for classification of sequence variation have been developed, which combine independent estimators to aid in characterization of sequence variation. Both independent estimators and combined risks for modeling systems may be integrated into a rigorous classification procedure that is based on clinical observation. This multidisciplinary approach will prevent over-reliance on one source for determination of causality of sequence variation and will allow for accurate interpretation of the potential causality of sequence variation. It is recognized that clinical diagnostic laboratories may not have access to the resources necessary to definitively resolve genotype/phenotype correlations. However, laboratories might consider establishing collaborative relationships with research facilities to determine genotype/phenotype correlations.
IV. AMENDED REPORTS: REPORTING UPDATED RESULTS TO PHYSICIANS
Clinical molecular genetics is a fast-moving field. Almost without exception, what is considered state of the art or state of knowledge today will not be the same in the future, sometimes even in the near future. As described above, the reporting of novel sequence variants to physicians and patients must include a clinical interpretation based on the best data available at the time of testing. Often, as subsequent studies are done, either within the same family or others reported in the literature, this best-guess clinical interpretation may need to be modified or changed. A suspected benign polymorphism may be confirmed as such by larger studies, or conversely it may be demonstrated to in fact represent a deleterious mutation. The reverse may occur for a variant that was initially designated a mutation. Although past statements by ACMG19 have stressed the impracticality of relocating and recontacting prior patients to inform them of the continuing advancements in the field relevant to their disorder and have largely placed the responsibility for monitoring new discoveries and treatments on the patients themselves and their primary care physicians, the issues surrounding novel sequence variants are somewhat different. Unlike major advancements involving more common conditions and mutations, these variants are by definition rare, and knowledge of them is often restricted to one or a few laboratories and may never make it into the published literature. The testing laboratory (with input from colleagues or the literature) may be in the best position to modify previously issued interpretations and should make an effort to contact physicians of previously tested patients in the event that new information changes the initial clinical interpretation of their sequence variant.
V. CLINICAL AND TECHNICAL VALIDATION OF SEQUENCE VARIATION DETECTION
Sequence analysis for clinical service may be provided only by laboratories holding current CLIA20 licensure. Clinical laboratories may send out sequencing tests to a CLIA-certified core facility. The ACMG recommends that interpretation and reporting of clinical molecular genetic data be limited to qualified providers such as those certified in the medical specialty of clinical molecular genetics by the American Board of Medical Genetics or equivalent.5 Tests must be accepted as clinically useful (clinical validation) and must be technically validated by the service provider.
References
Kazazian HH, Boehm CD, Seltzer WK . ACMG recommendation for standards for interpretation of sequence variations. Genet Med 2000; 2: 302–303.
Human genome mutation database at the Institute of Medical Genetics in Cardiff Wales United Kingdom: Cardiff University. 2004 Available at: http://archive.uwcm.ac.uk/uwcm/mg/hgmd0.html Accessed November 26, 2007.
db SNP at the National Center for Biotechnology Information in Bethesda, Maryland, 1988. Available at: www.ncbi.nlm.nih.gov/sites/entrez?db=snp Accessed November 26, 2007.
Fitze G, Schreiber M, Kuhlisch E, Schackert HK, et al. Association of RET proto-oncogene codon 45 with Hirschsprung Disease. Am J Hum Genet 1999; 65: 1469–1473.
American College of Medical Genetics standards and guidelines for clinical genetics laboratories, 2007 edition. Available at: http://www.acmg.net/AM/Template.cfm?Section=Laboratory_Standards_and_Guidelines&Template=/CM/HTMLDisplay.cfm&ContentID=2009 Accessed November 26, 2007.
College of American Pathologists molecular pathology checklist. Available at: www.cap.org/apps/docs/laboratory_accreditation/checklists/molecular_pathology_december2006.pdf Accessed November 26, 2007.
den Dunnen JT, Antonarakis SE . Nomenclature for the description of human sequence variations. Hum Genet 2001; 109: 121–124.
den Dunnen JT, Antonarakis SE . Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 2000; 15: 7–12.
den Dunnen JT. Nomenclature for the description of sequence variations. Revised October, 2007, on the Human Genome Variation Society (HGVS). Available at: http://www.hgvvs.org/mutnomen/ Accessed November 26, 2007.
NCBI refseq database at the National Center for Biotechnology Information in Bethesda, Maryland. Available at: http://www.ncbi.nlm.nih.gov/RefSeq/ Accessed November 28, 2007.
Human Variome Project, 2007. Available at: http://www.humanvariomeproject.org/ Accessed November 28, 2007.
Health Insurance Portability and Accountability Act of 1996 (HIPAA) at U.S. Department of Health and Human Services at Washington, D.C. Available at: http://www.hhs.gov/ocr/hipaa/. Accessed November 28, 2007.
Grantham R . Amino acid difference formula to help explain protein evolution. Science 1974; 185: 862–864.
Ng PC, Henikoff S . SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 2003; 31: 3812–3814. Available at: http://blocks.fhcrc.org/sift/SIFT.html Accessed November 28, 2007.
Polyphen (polymorphism, phenotyping) at European Molecular Biology Laboratory, Meyerhofstrasse 1, D-69117 Heidelberg, Germany, 2002. Available at: http://coot.embl.de/PolyPhen/ Accessed November 28, 2007.
Berezin C, Glaser F, Rosenberg Y, Paz I, et al. ConSeq: the identification of functionally and structurally important residues in protein sequences. Bioinformatics 2004; 20: 1322–1324. Available at: http://conseq.bioinfo.tau.ac.il/ Accessed November 28, 2007.
Splice Site Neural Network at the University of California, Berkeley, California, 1997. Available at: http://www.fruitfly.org/seq_tools/splice.html Accessed November 28, 2007.
ESEfinder:Exonic Splicing Enhancer Finder at Cold Spring Harbor Laboratory, Cold Spring Harbor, 2001. Available at: http://rulai.cshl.edu/tools/ESE/index.html Accessed November 28, 2007.
Social, Ethical and Legal Issues Committee ACMG policy statement: duty to recontact. Genet Med 1999; 1: 171–172.
Clinical Laboratory Improvement Amendment (CLIA) at the U.S. Department of Health and Human Services, Baltimore, Maryland. Available at: http://www.cms.hhs.gov/clia/. Accessed November 28, 2007.
Acknowledgements
The authors thank Dr. Richard Cotton for helpful review of this manuscript. The revised document was reviewed and endorsed by the Molecular Working Group of the ACMG Quality Assurance Committee.
Author information
Authors and Affiliations
Consortia
Additional information
Disclaimer: These Technical Standards and Guidelines were developed primarily as an educational resource for clinical laboratory geneticists to help them provide quality clinical laboratory genetic services. Adherence to these standards and guidelines is voluntary and does not necessarily assure a successful medical outcome. These Standards and Guidelines should not be considered inclusive of all proper procedures and tests or exclusive of other procedures and tests that are reasonably directed to obtaining the same results. In determining the propriety of any specific procedure or test, the clinical laboratory geneticist should apply his or her own professional judgment to the specific circumstances presented by the individual patient or specimen. Clinical laboratory geneticists are encouraged to document in the patient's record the rationale for the use of a particular procedure or test, whether or not it is in conformance with these Standards and Guidelines. They also are advised to take notice of the date any particular standard or guidelines was adopted, and to consider other relevant medical and scientific information that becomes available after that date.Sue Richards, PhD, Molecular/Medical Genetics, MP-350, DNA Lab, Oregon Health & Science University, 3181 SW Sam Jackson Park Road, Portland OR 97239. E-mail: richarsu@ohsu.edu.
Rights and permissions
About this article
Cite this article
Richards, C., Bale, S., Bellissimo, D. et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med 10, 294–300 (2008). https://doi.org/10.1097/GIM.0b013e31816b5cae
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e31816b5cae
Keywords
This article is cited by
-
Novel homozygous frameshift variant in the ATCAY gene in an Iranian patient with Cayman cerebellar ataxia; expanding the neuroimaging and clinical features: a case report
BMC Medical Genomics (2023)
-
Next-generation sequencing of 500 POI patients identified novel responsible monogenic and oligogenic variants
Journal of Ovarian Research (2023)
-
Identification of BRCA1/2 mutation female carriers using circulating microRNA profiles
Nature Communications (2023)
-
A presumed missense variant in the U2AF2 gene causes exon skipping in neurodevelopmental diseases
Journal of Human Genetics (2023)
-
Genetic analysis assists diagnosis of clinical systemic disease in children with excessive hyperopia
BMC Pediatrics (2022)
