
Abstract
Purpose: X-linked infantile spinal-muscular atrophy (XL-SMA) is a rare disorder, which presents with the clinical characteristics of hypotonia, areflexia, and multiple congenital contractures (arthrogryposis) associated with loss of anterior horn cells and death in infancy. We have previously reported a single family with XL-SMA that mapped to Xp11.3-q11.2. Here we report further clinical description of XL-SMA plus an additional seven unrelated (XL-SMA) families from North America and Europe that show linkage data consistent with the same region.
Methods: We first investigated linkage to the candidate disease gene region using microsatellite repeat markers. We further saturated the candidate disease gene region using polymorphic microsatellite repeat markers and single nucleotide polymorphisms in an effort to narrow the critical region. Two-point and multipoint linkage analysis was performed using the Allegro software package.
Results: Linkage analysis of all XL-SMA families displayed linkage consistent with the original XL-SMA region.
Conclusion: The addition of new families and new markers has narrowed the disease gene interval for a XL-SMA locus between SNP FLJ22843 near marker DXS 8080 and SNP ARHGEF9 which is near DXS7132 (Xp11.3-Xq11.1).
Similar content being viewed by others


Main
X-linked infantile spinal muscular atrophy (MIM 301830;OMIM: Online Mendelian Inheritance in Man at http://www3.ncbi.nlm.nih.gov/) is a rare congenital disorder characterized by multiple joint contractures. Hall et al.2 distinguished three different types of X-linked spinal muscular atrophy: 1) a severe (lethal) type, characterized by severe contractures, scoliosis, chest deformities, hypotonia, and death due to respiratory insufficiency within three months of birth; 2) a moderately severe type, characterized by severe contractures, ptosis, microphallus, cryptorchidism, inguinal hernias, and normal intelligence; and 3) a resolving type, characterized by mild to moderate contractures at birth that improve with time. Progressive loss of anterior horn cells was cited as the cause of the severe lethal form of X-linked spinal muscular atrophy. Greenberg et al.3 described under the label “X-linked infantile spinal muscular atrophy” a similar disorder to X-linked spinal muscular atrophy Type I2 that appeared to be X-linked recessive and was associated with hypotonia, areflexia, chest deformities, facial dysmorphic features, and congenital joint contractures (arthrogryposis). The findings of electromyography and muscle biopsy were consistent with autosomal recessive infantile spinal muscular atrophy (Werdnig-Hoffmann, MIM 253300).
We have previously described meiotic breakpoint mapping (concordance analysis), and whole chromosome multipoint linkage analysis to the X-linked spinal muscular atrophy family reported by Greenberg et al.3 We showed localization of the disease gene to a pericentromeric interval from Xpll.3-qll.2 (Zmax = 2.63; between MAOB and the marker DXS9911). In this report, we extend our previous clinical and molecular investigations to eight new unrelated families with disease presentation and inheritance similar to that described by Greenberg et al.3
MATERIALS AND METHODS
X-linked SMA patients and DNA isolation
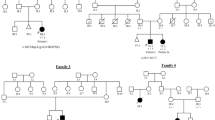
All human subject activities occurred in compliance with an active human subjects protocol, which has institutional approval. Patients were identified and recruited nationally and internationally through a “Call for Patients” (published in selected scientific journals), through scientific presentations at regional, national/international meetings, and through sources of clinical collaborations. The medical and clinical histories of all potential XL-SMA cases were discussed by Dr. Baumbach and referring physician before acquisition. Once a patient was deemed to meet study inclusion criteria, recruitment and consent were obtained by standard procedures. Recruitment of children and minors was approved in the human subjects protocol mentioned above and as such, adhered to institutional and national ethical standards for inclusion of children in clinical research. Similar statements apply to the inclusion of women and minorities into this study. All families (Table 1) were ascertained based on affected male probands showing: clinical findings of severe hypotonia (Table 2; Fig. 1), contractures and/or fractures at birth, or shortly thereafter; muscle biopsy indicative of neurogenic atrophy involving both fiber types; histology consistent with Werdnig-Hoffmann; EMG indicative of denervation; autopsy examination (when available) indicating loss of anterior horn cells and other histological and/or pathologic findings consistent with Werdnig Hoffmann; and death within the first two years of life due to respiratory distress or pulmonary hypoplasia. Other associated features included undescended testes, hypospadias, or other genital abnormalities in at least one affected male per family; family history of spontaneous abortions, miscarriages, decreased fetal movements, or Caesarian section deliveries; and dysmorphic features (skull and faces) in affected males at birth. Genomic DNA was isolated either from whole blood collected in EDTA tubes or transformed lymphoblastoid cell lines as previously described.4 In addition, affected patients who were ascertained prospectively were tested for SMN1 mutations, and were found to be negative prior to linkage testing.

Representative patient photos: (A) Proband of Family 2 at birth; (B) proband at eight month of age, shortly before his death.
Genotyping
Primer sequences for microsatellites were obtained from the Genome Data Base. Genotyping of all available family members was completed either using fluorescent multiplex PCR primers analyzed using an ABI (Applied Biosystems, Foster City, CA) automated DNA sequencer5,6 or the incorporation of radioactive label during PCR amplification of DNA markers. Primers for radioactive genotyping were purchased from Research Genetics, Inc., and the forward primers were labeled with γ-ATP using polynucleotide kinase (PNK) from New England Biolabs. PCR was performed on DNA samples with a first cycle consisting of a 10-minute denaturation at 94°C followed by 30 cycles of 94°C for 25 seconds, 56°C for 25 seconds, and 72°C for 1 minute. Equal volumes of PCR reaction and loading buffer (98% formamide, 10mM EDTA, 3mg/mL bromophenol blue, and 3mg/mL xylene cyanol) were mixed and the samples were then denatured at 94°C for 5 minutes and were immediately placed on ice. Three microliters were loaded on a 6% polyacrylamide denaturing gel and electrophoresed for 3–5 hours at 1500 V. Gels were transferred to filter paper and exposed to X-ray film.
Linkage studies were completed using Families 1, 2, 4, 5, 6, 7 and 12 in one of the two senior authors' laboratories (L.B.R., Miami, or E.P.H., Washington, D.C.). These results were then independently analyzed by the other laboratory, with mutual agreement derived to achieve final genotype data used for LOD score calculations. For Family 3, linkage experiments were performed in Spain by F.M. and F.P.
We then saturated the candidate gene region, in order to detect recombination events and to further narrow the candidate gene interval in the three largest pedigrees, Families 2, 3, and 5. These studies used a panel of 15 microsatellite repeat markers and 14 Single Nucleotide Polymorphisms (SNPs). The methodology for these markers and SNPs used microsatellite repeat markers from the Applied Biosystems Linkage Mapping Set and the Applied Biosystems catalogue of Assay-on-Demand SNPs.
Each marker is a dinucleotide repeat selected from the Genethon human linkage map. PCR reactions are run on the ABI 3100 Genetic Analyzer and allele discrimination is determined using ABI GeneMapper software. SNP reactions are performed and analyzed using an ABI 7000 Sequence Detection System.
The Allegro software package was used for all two-point and multipoint linkage analyses. XL-SMA was assumed to be X-linked recessive, completely penetrant, with a disease gene frequency of 1 × 10−4. Allele frequencies were generated by allele counting in unrelated individuals, and relative map positions for the markers used in multipoint linkage analyses were based on data from Genome Data Base (http://gdbwww.gdb.org/), Research Genetics, Inc. (www.resgen.com/), and Cedar Genetics (cedar.genetics.soton.ac.uk/public_html/). We have tested all of the families in this region for homogeneity using the Homog program version 3.35 and failed to reject the null hypothesis of homogeneity (P = 1), indicating that our data are consistent with homogeneity.
RESULTS
Clinical data
We have described an X-linked recessive form of infantile lethal motor neuron disease (MIM 301830), which closely resembles Werdnig-Hoffmann disease, with additional differentiating features including early onset or congenital contractures and/or fractures.1 Based on diagnostic criteria and a strong positive family history a subset of six additional unrelated families were selected for this study (Table 1). All affected males in these pedigrees displayed a disease course similar to Type I Proximal Spinal Muscular Atrophy (PMSA) with mortality in 75% of patients within the first two years of life (Table 2). All families included in this study have had at least one affected male die within the first year of life. Additionally, several of the families (Families 1, 2, and 5) had an affected male who, with critical medical intervention, survived into childhood. Muscle biopsies and/or EMG studies were consistent with neurogenic atrophy, and autopsy examination in seven cases showed loss of anterior horn cells in affected males. Other associated clinical features (congenital or early-onset proximal and digital contractures, facial weakness and other facial dysmorphic features, and urogenital abnormalities) have been noted in several affected males (data not shown). Obligate carrier females showed no clinical manifestations of the disorder.
Linkage studies
Linkage studies were conducted in seven unrelated multi-generational XL-SMA families (Table 1) using microsatellite repeat markers from the candidate disease gene region, Xp11.3-q11.2, previously reported by our group.1 Families 8–11 were not large enough for linkage analysis. Concordance analysis was used to define maternal meiotic recombination breakpoints surrounding a disease gene region – this was followed by multipoint linkage analysis using a high density of DNA markers in the candidate region.
Families 1, 4, 5, 6, and 7 were preliminarily investigated in the U.S. (L.B.R., Miami, or E.P.H., Washington, D.C.) using three of the markers that had been previously shown to demonstrate positive linkage in Family 2 (DXS988, DXS991 and DXS1003).1 These preliminary linkage studies were completed for each family, and when linkage to this region was confirmed, families were further examined with additional markers from within this region, generating two-point LOD score data for each family (data not included). LOD score calculations for 5/6 individual families (Families 1, 2, 5, 6, 7) were added to generate two-point LOD score for each of the markers spanning the Xp11.4-Xq11.1 region (Table 3). The highest two-point LOD score was found with DXS7132 (Zmax = 4.43; θ = 0.00).
The combined multipoint LOD scores across the centromeric region of the X-chromosome are displayed in Table 4 and Figure 2. The overlapping multipoint linkage analyses showed significantly positive LOD scores in the region between DXS8035 (Xp11.3) and DXS1194 (Xq11.1; see Fig. 2). Families 1, 2, 5, 6 and 7, showed a maximum multipoint LOD score of 6.72 and 6.75 at DXS991 and DXS1190, respectively, representing a smaller positive LOD score interval. In addition to the families analyzed above, two of the coauthors (F.M. and F.P.) determined positive linkage to the same region (between the DMD locus (Xp22.1) and DXS983 (Xq12) in the XL-SMA family they contributed (Family 3, Table 1), while also excluding the remainder of the X-chromosome. Multipoint linkage analysis was also conducted on Family 3, which demonstrated a maximum LOD score of 2.12 at DXS1003 (θ = 0.00; Table 4). The region of the X-chromosome linked in this family is larger than that previously defined by our group,1 due to choice of DNA markers used in these studies. With the addition of the LOD score data for family 3 at DXS1003 (see Table 4) to the LOD scores of Families 1, 2, 5, 6 and 7, the cumulative maximum LOD score is at DXS1003 (Zmax = 8.71; θ = 0.00).

Multipoint linkage analysis covering the X chromosome from Xp11.3 to Xq11.1: Four-point analyses were calculated using Allegro. Only the region defined by markers DXS8035 and DXS1194 showed positive LOD scores.
Not included in any LOD score calculations are the results of linkage analysis completed on family 12. We are designating this family as one presently undetermined. There is some evidence that this family may represent a new mutation, however, until we have isolated the gene responsible for this disease and can conduct mutation analysis we cannot absolutely state this. The evidence of this family possibly representing a new mutation is that current haplotype data are highly suggestive of linkage to the XL-SMA region: the two affected boys share the same haplotype, and their unaffected uncle has a different haplotype. Additionally, when this family is analyzed with a mutation frequency of 1 × 10−4 for both male and female meioses there is positive linkage to this region with a LOD score of 0.5 (data not shown). We might expect that for an X-linked lethal recessive disorder (e.g. Duchenne Muscular Dystrophy) that some proportion of the cases represent new mutational events.
In order to narrow the XL-SMA candidate region, we focused our efforts on the use of new polymorphic markers between MAOB and DXS1003 and distally, between DXS1194 and the centromere to better identify proximal and distal breakpoints. Through detailed genetic examination of XL-SMA family 2 (the largest of the families) using these new markers, we were able to detect two independent recombination events that eliminated markers DXS8035 and DXS1194 (Fig. 4). No recombinant X chromosomes involving crossovers in the Xp11.3-q11.1 were observed in any other of our families.

DNA linkage studies in of XL-SMA Family 2 reveals recombination events: The minimum disease haplotype for XL-SMA is indicated by the box. Affected individuals are indicated by blackened symbols, carrier females are indicated by circle containing a black circle, and unaffected individuals are indicated by unblackened symbols. All affected males and carrier females share the disease haplotype. DNA recombination occurred from III:4 to IV:3 eliminating marker DXS8035. Recombination also occurred from II:3 to III:7 eliminating marker DXS1194. Arrows indicate these events.
We further refined the candidate gene interval by studying 15 microsatellite repeat markers and 14 SNPs in the three largest familes (Families 2, 3, and 5). In families 2 and 5 we were able to include additional family members that were not previously available for study. This analysis revealed a recombination event in an affected male in Family 2 which allows us to eliminate the NDP gene (Fig. 5). There are also two recombination events in noncarrier females which are not informative for XL-SMA, one in Family 2 and one in Family 5 (Fig. 5 and 6).

SNP analysis of Family 2: SNP analysis in this family shows a recombination in individual IV:2, an affected male, between NDP and FLJ22843, which allows elimination of the NDP gene. There is a second recombination in a noncarrier female, individual III:15, which is not informative for XL-SMA.

SNP analysis of Family 5: SNP analysis in this family shows a recombination in individuals III:8 and III:9 between GRIPAP1 (Xp11.23) FLJ31204 (Xp11.21); however this does not help to narrow the region as the recombination has occurred in a portion of the family without affected individuals.
Additionally, with the data from these 15 markers and 14 SNPs we were able to calculate the combined multipoint LOD scores across the centromeric region of the X-chromosome. (Table 5; Fig. 3). These data showed significantly positive LOD scores in the region between the SNP FLJ22843 (located close to DXS8080) (Xp11.3) and SNP ARHGEF9 (located near marker DXS7132) (Xq11.1; Fig. 3). Families 2, 3, and 5, showed a maximum multipoint LOD score of 4.0818 and 4.0766 at UTX and DXS8026, respectively. The region from FLJ22843 to DXS8023 showed a LOD score of 3.599 to 3.75 this suggests a somewhat smaller region of peak LOD scores than the earlier analysis but is only based on the three largest families.

Linkage plot of XL-SMA Families 2, 3, and 5: Additional microsatellite markers and SNPs analyzed in the three largest XL-SMA families confirm and narrow the candidate gene region.
Therefore, all XL-SMA families identified and studied by linkage analysis are linked to the same X-chromosome region – there is no evidence of genetic heterogeneity at this time. There is no evidence for a shared haplotype between the families.
DISCUSSION
Based on the study of eight unrelated families (families 1–7, and 12) from Europe, Mexico and the United States, this report provides important new evidence for linkage of an X-linked infantile spinal muscular atrophy locus to Xp11.3-Xq11.1. Unlike classical SMA congenital contractures have been associated with each of these cases.
Arthrogryposis involves multiple congenital contractures and limited movement of body areas, more often distal than proximal.7 It is considered to be a rare birth defect and lethal in one-third of all cases. The primary cause is believed to be decreased fetal movement7,8 while other major causes include neuropathic and myopathic disorders, congenital abnormalities of connective tissues and joints, and physical causes. A neurogenic basis has long been noted, and is listed as a distinct entity listed in OMIM (MIM 208100). Neuropathic causes of lethal arthrogyposis include absent, abnormal, or nonfunctional nerves in both the central and peripheral nervous systems.7,8 Since the 1980s, a growing literature has documented a strong association between anterior horn cell loss and arthrogyposis.7,8
Other related clinical syndromes include Pena-Shokier, (MIM 208150), Lethal Contracture Syndrome (MIM 253310) and Congenital Benign SMA with Lower Extremity Contractures (MIM 600175). A significant number of these reported cases have had clinical investigations, muscle biopsies and/or autopsy studies consistent with neurogenic atrophy/anterior horn cell loss (MIM 208100). Thus, a growing body of evidence supports a strong association, if not causal, between congenital anterior horn cell loss and forms of arthrogryposis. Mutations in the SMN1 gene have been documented in some cases,9 while in others, linkage to the SMN region has been excluded,9,10 further supporting the existence of additional unidentified genes in the human genome involved in the maturation, preservation and selective death of anterior horn cells. It is of interest that arthrogyropsis was an exclusion criterion for the 5q SMA gene localization studies.11,12
The existence of a distinct X-linked syndrome, similar to SMA Type I in association with congenital contractures (and fractures), has long been suggested. As early as 1938,13 sporadic case reports of male infants with congenital contractures and a SMA Type I phenotype were published. (See14–18 for review). Several landmark papers in the last 20 years recognized the existence of this previously under recognized syndrome. Two of these have been previously discussed.2,3 A second multigenerational severe X-linked spinal muscular atrophy pedigree was reported almost a decade later.19 However, neurogenic atrophy was not noted, indicating clinical heterogeneity even within forms of XL-SMA. In 1991, Borochowitz14 reported two male siblings with a syndrome almost identical to that described by Greenberg et al3.
Mapping of rare X-linked recessive disorders with a poor prognosis is often difficult because of a limited availability of samples from deceased affected family members. We continued to use meiotic breakpoint mapping (concordance analysis) in addition to two-point and multipoint linkage analysis to analyze the families in this cohort. As a result of these investigations, seven additional families were tested for linkage on the X-chromosome; six of the seven multigenerational families were useful for generating meaningful LOD scores, and all families mapped to the same region as the first family reported by our group.1 The maximum cumulative LOD score for the seven linked multi-generational families occurs at DXS1003 (Zmax = 8.71 at θ = 0.0).
Multipoint linkage analysis using the five families identified recombination events, and narrowed the candidate disease gene interval to a region defined by DXS8035-DXS1194 (Fig. 4), limiting the region to 21 cM or 21.6 Mb encoding approximately 200 genes. The disease gene interval being in such close proximity to the centromere has presumably hampered our efforts to find additional recombination events, because recombination frequency is very low close to the centromere.20,21
These results strongly support the existence of a disease locus for XL-SMA within this region.
Analysis of linkage data from only families 2, 3, and 5 using both microsatellite markers and SNPs generated a maximum multipoint LOD score of 4.0818 and 4.0766 at UTX and DXS8026, respectively, suggesting a slightly smaller region of peak LOD scores than that suggested by analysis of the five families. The overall congruence of positive LOD score in essentially the same region of the X chromosome by data generated by different laboratory methods and separate LOD score analysis lends further evidence that the disease-causing gene for X-linked SMA lies within this region.
Characterizing the gene defect in X-linked SMA will shed important new light on how the underlying genetic defect leads to diverse congenital anomalies and neuromuscular manifestations. Most importantly perhaps, our data provide the basis for isolation of the gene for an X-linked lethal lower motor neuron disease. Identification and characterization of this gene will most likely reveal an essential component of the motor neuron system and provide new insight into human motor neuron disease.
It is clear that spinal muscular atrophy is associated with neuronal apoptosis of the lower motor neurons. Approximately 4% of SMA patients failed to show mutations within the SMN1 gene.22 Our data suggests that XL-SMA may not be as rare as previously assumed, and that patients testing negative for SMN1 mutations might instead be affected by XL-SMA. The discovery of this gene will provide major insights into these issues, and eventually into the etiology and developmental timing of motor neuron loss.
References
Kobayashi H, Baumbach L, Matise T, Schiavi A, et al. A gene for a severe lethal form of X-linked arthrogryposis (X-linked infantile spinal muscular atrophy) maps to human chromosome Xp11.3-q11.2. Hum Mol Genet 1995; 4: 1213–1216.
Hall JG, Reed SD, Scott CI, Rogers JG, et al. Three distinct types of X-linked arthrogryposis seen in 6 families. Clin Genet 1982; 21: 81–97.
Greenberg F, Fenolio KR, Hejtmancik JF, Armstrong D, et al. X-linked infantile spinal muscular atrophy. Am J of Disease Child 1988; 142: 217–219.
Miller SA, Dyles DD, Plesky HF . A simple salting our procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Kobayashi H, Matise TC, Marks HG, Perlin MW, et al. Towards fully automated genotyping: use of an X linked recessive spastic paraplegia family to test alternative analysis methods. Hum Genet 1995; 95: 83–90.
Pegoraro E, Schimke RN, Arahata K, Hayashi Y, et al. Detection of new paternal dystrophin gene mutations in isolated cases of dystrophinopathy in females. Am J Hum Genet 1994; 54: 989–1003.
Banker BQ . Congenital Deformities Myology, Vol II. New York: McGraw-Hill. 1985.
Hall JG . Genetic aspects of arthrogryposis. Clin Orthop 1985; 194: 44–53.
Burglen L, Amiel J, Viollet L, Lefebvre S, et al. Survival motor neuron gene deletion in the arthrogryposis multiplex congenita-spinal muscular atrophy association. J Clin Invest 1996; 8: 1130–1132.
Vuopala K, Makela-Bengs P, Suomalainen A, et al. Lethal congenital contracture syndrome (LCCS), a fetal anterior horn cell disease, is not linked to the SMA 5q locus. J Med Genet 1995; 32: 36–38.
Dubowitz V . Chaos in classification of the spinal muscular atrophies of childhood. Neuromuscul Disord 1991; 1: 77–80.
Munsat TL . Workshop Report of the SMA International Working Collaborative Group. Neuromuscul Disord 1991; 1: 81.
Stoeber E . Kleine Mitteilung: Uber atonisch-sklerotische muskeldystrophie (Typ Ullrich). Z. Kinderheilkunde 1938; 60: 279–284.
Borochowitz Z, Glick B, Blazer S . Infantile spinal muscular atrophy (SMA) and multiple congenital bone fractures in sibs: a lethal new syndrome. J Med Genet 1991; 28: 345–348.
Fahy MJ, Hall JG . A retrospective study of pregnancy complications among 828 cases of arthrogryposis. Genet Couns 1990; 1: 3–11.
Miller A, Solimano A, Norman MG . Arthrogryposis multiplex congenita and hypotonia in a male neonate. Pediatr Neurosci 1987; 13: 272–277.
Hageman G, Hoogenraad TU, Prevo RL . The association of cortical dysplasia and anterior horn arthrogryposis: a case report. Brain Dev 1994; 16: 463–466.
Vogel H, Halpert D, Horoupian DS . Hypoplasia of posterior spinal roots and dorsal spinal tracts with arthrogryposis multiplex congenita. Acta Neuropathol 1990; 79: 692–696.
Hennekam RC, Barth PG, Van Lookeren Campagne W, et al. A family with severe X-linked arthrogryposis. Eur J Pediatr 1991; 150: 656–660.
Nagaraja R, MacMillan S, Kere J, Jones C, et al. X chromosome map at 75-kb STS resolution, revealing extremes of recombination and GC content. Genome Res 1997; 7: 210–222.
Payseur BA, Nachman MW . Microsatellite variation and recombination rate in the human genome. Genetics 2000; 156: 1285–1298.
Wirth B . Spinal muscular atrophy (SMA): State-of-the art and therapeutic perspectives. ALS and other motor neuron disorders 2002; 3: 87–95.
Acknowledgements
This study has been generously supported by grants from the Muscular Dystrophy Association, the Families of SMA, the Patterson Trust, the Pentland-Hall Foundation of Dade County, the Spanish Ministry of Science and Technology and the Fondo de Investigación Sanitaria. The scientific team is indebted to the numerous collaborating clinicians, geneticists and other scientists, as well as the XL-SMA families, who have significantly contributed their time, efforts, and expertise to this project over the years. DCD was supported by a pre-doctoral fellowship award from the American Heart Association.
Author information
Authors and Affiliations
Corresponding author
Additional information
D.D. and M.E.A. contributed equally.
Rights and permissions
About this article
Cite this article
Dressman, D., Ahearn, M., Yariz, K. et al. X-linked infantile spinal muscular atrophy: Clinical definition and molecular mapping. Genet Med 9, 52–60 (2007). https://doi.org/10.1097/GIM.0b013e31802d8353
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e31802d8353
Keywords
This article is cited by
-
The Genetics of Spinal Muscular Atrophy: Progress and Challenges
Neurotherapeutics (2015)
-
Type 0 spinal muscular atrophy with multisystem involvement
Indian Pediatrics (2014)
