
Abstract
Purpose: Humans with small deletions of chromosome 21 provide important models for understanding the role of dosage-sensitive genes in brain morphogenesis. To identify chromosome 21 genes responsible for defects of the central nervous system, we determined the deleted regions and brain malformations in three unrelated individuals with overlapping partial deletions of chromosome 21.
Methods: Fluorescent in situ hybridization and magnetic resonance imaging were used to define the chromosomal structure and structural brain abnormalities present in these three individuals.
Results: The regions of chromosome 21 found to be deleted in these individuals were as follows: case 1: KCNJ6 to the telomere; case 2: ITSN1 to the telomere; and case 3: ITSN1 to PCNT2. The abnormalities of brain structure shared by all included microcephaly, pachygyria, polymicrogyria, colpocephaly, hypoplastic corpus callosum and white matter, hypoplastic cerebellum, and enlarged ventricular system. The clinical features in common included mental retardation, microcephaly, facial dysmorphism, and epilepsy (severe in one patient).
Conclusion: From analyses of the molecular, cytogenetic, and neuroimaging data from these three individuals, combined with those from previously reported cases, we infer that deletion of an 8.4-Mb region in chromosome band 21q22.2-22.3 (KCNJ6-COL6A2) is associated with cortical dysplasia. We propose that one or more dosage-sensitive genes in this region contributes to cortical development and that deletion of 21q22.2-22.3 should be considered in the diagnosis of mentally retarded patients with facial dysmorphism and cerebral dysplasia.
Similar content being viewed by others


Main
Down syndrome (DS), usually caused by trisomy for human chromosome 21 (HSA 21), is a major cause of mental retardation and is characterized by abnormalities of cortical neuroanatomy, neurochemistry, and function.1–3 In comparison, the brain abnormalities associated with chromosome 21 deletions are much more severe. Individuals with small deletions of HSA 21 may thus provide an important model to link specific chromosomal regions and genes to structural brain malformations, an approach that may provide the means to understand the role of subtle variations in gene expression in normal variation.
The association of an HSA 21 interval with a given phenotype when duplicated or deleted remains an important first step in identifying the chromosome 21 genes and molecular pathways underlying the pathogenesis of DS. The complete nucleotide sequence of HSA 21 has been reported,4 and 225 genes detectable using specific markers and clones were subsequently annotated.5 Currently, the Ensembl database lists 243 known genes and 87 pseudogenes on HSA 21 (http://www.ensembl.org). There are several reports linking specific phenotypes with partial monosomy or trisomy 21,6–11 and these have been used to generate a phenotypic map of 25 DS features.12 However, despite extensive analyses of gene expression patterns and data from overexpressing mouse models, there are as yet few definitive links between individual HSA 21 genes and the phenotypes of DS. Moreover, of the few reports of human gene mutations that disturb cortical development, none are located on HSA 21. Identifying HSA 21 genes involved in human brain morphogenesis would be useful for understanding normal development and the origins of abnormal brain structures in DS.
We now report neuroimaging analysis of brain malformations in three individuals with partial deletions of HSA 21. Combining these data with the results of our cytogenetic and molecular studies, and with the data from previously published individuals with partial monosomy 21,13–15 suggests that an 8.4-Mb region of 21q22.2-22.3 (KCNJ6-COL6A2) is associated with cortical dysplasia, specifically pachygyria (few broad, flat gyri and shallow sulci), polymicrogyria (too many abnormally small brain convolutions), and colpocephaly (abnormal enlargement of the occipital horns of the brain). We propose that one or more dosage-sensitive genes located in this region contribute to cortical development, and that 21q22.3 deletion should be considered in the diagnosis of mentally retarded patients with facial dysmorphism and cerebral dysplasia.
MATERIALS AND METHODS
Clinical analyses
The clinical features in three individuals with partial monosomy 21 were assessed as indicated in Table 1. These data were derived from original records of the primary care physicians, the examining geneticists, and the consulting specialists. Full institutional review board-approved procedures were followed, and confidentiality was maintained throughout. The ocular findings will be considered in a future publication.
Cytogenetic analysis
Extended chromosome preparations were prepared from cultured peripheral lymphocytes by methotrexate synchronization.16 Chromosomes were stained by G-banding (GTG) and R-banding (RHG) techniques.16
Molecular analysis
A series of previously mapped17–20 chromosome 21 DNA markers were used as probes to define the copy number and/or structural rearrangement in individuals with partial monosomy 21. The sources and references for each of these probes can be found in Table 2. DNA probes were extracted from BAC, PAC, or cosmid clones and labeled by indirect or direct methods using a Nick Translation Kit (Invitrogen, Carlsbad, CA). Biotin-11-dUTP or Dig-11-dUTP (Sigma, St. Louis, MO) were used for indirect labeling, and Alexa Fluor 488-5-dUTP, Alexa Fluor 568-5-dUTP, Alexa Fluor 594-5-dUTP, and Alexa Fluor 647-5-dCTP (Molecular Probes, Eugene, OR) were used for direct labeling.21 To define the breakpoints in each individual case, the strategy used was to perform an initial screen, followed by more specific testing with BACs progressively closer to the breakpoints.
Fluorescent in situ hybridization
Fluorescent in situ hybridization (FISH) and posthybridization detection were performed as previously described.21 Multicolor images were captured with a Zeiss (Munich, Germany) Axioplan 2 microscope equipped with an Axiocam MRM camera and conjugated to Metasystem software (Microsoft, Redmond, Washington). An example of four-color FISH showing the chromosome 21 deletion in case 1 is shown in Figure 1.

Four-color FISH illustrating the chromosome 21 deletion in case 1. One of the chromosomes 21 is deleted for BACs B855C1 and B74C9. B855C1 (red dot), B74C9 (yellow dot), B76H7 (pink dot), and B427H6 (green dot).
RESULTS
Case 1
This male child, now 5 years and 2 months of age, was delivered at 40 5/7 weeks gestation by cesarean section for fetal decelerations. The G3, P1 mother was 24 years old. Both parents appeared normal, and the family history was unremarkable. Dysmorphic features including facial abnormalities were noted immediately after birth, as well as low birth weight (2.065 kg) and microcephaly. Other clinical features are noted in Table 1. The ocular findings will be considered in a future publication. At 9 months of age, he developed intractable epileptic seizures that consisted of tonic and right partial seizures. These worsened with time and at 2 years of age were poorly controlled by medication. Clinical examinations now show severe developmental delay, altered gait associated with hypotonia, and mental retardation.
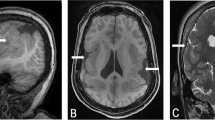
Magnetic resonance imaging (MRI) examinations performed at 1 day, 9 months, and 2 years of age revealed clearly abnormal brain morphology (Fig. 2,Table 3). At 2 years of age, MRI showed cortical dysplasia with areas of pachygyria, polymicrogyria, and colpocephaly. Paucity of cerebral white matter was most marked in the left parietal and occipital lobes. There was a slight increase in diffuse ventriculomegaly and widening of the sulci relative to the 9-month examination, likely reflecting a diffuse atrophy. Hypoplastic cerebellar vermis and corpus callosum were also detected, and the hippocampus was symmetrically malformed. Although there had been interval progression of the myelination of white matter in the cerebral hemispheres from the 9-month examination, persistent areas of T2 prolongation were observed in the subcortical white matter in the frontal and temporal lobes and insula, consistent with delayed myelination. The odontoid process was hypoplastic, and ossification was delayed. In all three cases, scans were viewed independently by at least two observers, author Laura Flores-Sarnat and Dr. Harvey Sarnat (Alberta Children's Hospital, Calgary, Canada), and by the referring neurologist.

MRI and computed tomography (CT) images of three cases with partial monosomy 21. A–D, MRI for case 1 at 2 years of age. A, Coronal MRI section showing polymicrogyria (thin arrow), left colpocephaly (thick arrow), and hypoplasia of vermis (arrowhead). B, Coronal section showing hypoplasia of the corpus callosum (thin arrow) and cortical dysplasia (thick arrow). C, Sagittal view showing occipital polymicrogyria (thin arrow). D, Sagittal view showing hypoplasia of the corpus callosum (thin arrow) and vermis of the cerebellum (thick arrow). E–H, MRI for case 2 at 1 year. E and F, Sagittal views show severe hypoplasia of the corpus callosum (thick arrow), occipital polymicrogyria (arrowhead), and a normal cerebellum and brain stem (thin arrow). G and H, Coronal sections showing cortical dysplasia, mainly pachygyria (thick arrow) and hypoplasia of the white matter (thin arrow). I–L, Cranial CT for case 3 at 2½ years of age (axial sections). I and J, Generalized cortical dysplasia. I, Polymicrogyria (thick arrow), hypoplasia of the corpus callosum (thin arrow), and atrophy of the cerebellum (arrowhead). J, Hydrocephalus ex vacuo (thin arrow). K and L, Hypoplasia of the white matter (thick arrow) and severe cortical dysplasia (thin arrow), more pronounced in the frontal and parietal than in the occipital areas.
Cytogenetic analysis revealed 46,XY,del21(q22.1). Molecular analysis was then performed by FISH, using subsets of 37 overlapping DNA probes extracted from BAC, PAC, or cosmid clones. These probes span over 27 Mb of chromosome 21 from RBM11 in 21q11.2 through the telomere and represent a total of 63 chromosome 21 markers and 56 genes (Table 2). This approach enables highly precise definition of the molecular breakpoints in each individual with partial aneuploidy 21. For case 1, a total of 19 probes spanning from D21S11 in 21q21.1 through the telomere (ter) were used. This individual was found to have deletions from KCNJ6 (P25A17) to ter (TelVysis 21), an 8.9-Mb region that includes 88 known genes (Figs. 1 and 3).

Summary of the chromosome 21 regions deleted in nine individuals with partial monosomy 21. Regions deleted in individuals with cortical dysplasia (black); regions deleted in individuals without cortical dysplasia (gray). Original references (superscripts) for previously reported cases. Minimal region of molecular overlap between the deletions in individuals with cortical dysplasia (black line with arrows at both ends). Locations of known genes mapping within the region are indicated. A full list of the genes mapping within the region can be found at http://www.ncbi.nlm.nih.gov/genome/guide/human/.
Case 2
This female child, now 15 years old, was born very small for gestational age with severe microcephaly, facial abnormalities, and a number of other dysmorphic features (Table 1). The family history was unremarkable. At 4 months of age she was hospitalized with mild respiratory distress and poor feeding, during which time she had two episodes of febrile seizures. MRI scans at 1 year of age showed multiple brain abnormalities (Fig. 2, Table 3), including diffuse cortical dysplasia with areas of pachygyria and polymicrogyria, hypoplasia of subcortical white matter and corpus callosum, and a normal cerebellum. The patient had severe global developmental delay and was profoundly mentally retarded.
Cytogenetic analysis revealed 46,XX,del21(q22). FISH was then performed as described for case 1, using a total of 12 DNA probes spanning from GART in 21q22.11 to ter (Table 2). This individual was found to have deletions from ITSN1 (T1276) to ter (TelVysis 21), a 12.87-Mb region that includes 104 known genes (Fig.3).
Case 3
This female child, now 15 years old, was born small for date at 41 weeks gestation to a 33-year-old G2, P1 mother. At birth she exhibited multiple abnormal features including microcephaly and dysmorphic facial features (Table 1). Head ultrasound revealed a hypoplastic corpus callosum and slight increase in size of the lateral ventricles, and a chest x-ray film revealed eventration of a hemidiaphragm. By 2 years of age she developed tonic-clonic seizures, confirmed by an electroencephalogram that showed paroxysmal activity originating in the left temporal lobe. A cranial CT scan at 2½ years of age revealed hydrocephalus ex vacuo and severe cortical dysplasia (affecting the left hemisphere more than the right), consisting of pachygyria, polymicrogyria, hypoplasia of the subcortical white matter and corpus callosum, and hypoplasia of the cerebellum (Fig. 2, Table 3).
Cytogenetic analysis demonstrated mosaicism with two different cell lines, 46,XX,del21(q22.2), unstable 21p11.1-ter (84%), and 46,XX (16%). FISH was performed using 22 DNA probes spanning from RBM11 in 21q11.2 to ter (Table 2). This individual was found to have deletions from ITSN1 (PF32E9) to PCNT2 (B78C7), a 12.6-Mb region that includes 102 known genes (Fig. 3).
DISCUSSION
Individuals with partial aneuploidy for HSA 21 provide rare opportunities to investigate the links between specific chromosomal regions, and possibly individual genes, and phenotypic features. In this report, we describe three individuals with overlapping deletions of chromosome 21 and structural brain malformations defined by MRI. Complete monosomy 21 results in death in utero, and the effects of partial monosomy 21 vary depending on the region deleted, size of the region, and presence of other chromosomal abnormalities. Although there are few reports in the literature of pure partial monosomy 21 6,8,13–15,22–24, there is evidence to suggest that deletion of the telomeric region D21S112-qter does not result in severe phenotypes.25,26 The brain abnormalities in the three individuals reported here include cortical dysplasia consisting of pachygyria, polymicrogyria and colpocephaly, hypoplasia of the corpus callosum and white matter, cerebellar hypoplasia, and enlargement of the ventricular system (Fig. 2). Although cortical dysgenesis has been described in human syndromes associated with deletions of chromosomes 17p13.327 and Xq22.3-q23,28 it has not been commonly reported in syndromes associated with deletions of chromosomes 15q11.2 (Prader-Willi), 5p15.2 (Cri-du-chat), 7q11.2 (Williams-Beuren), and 22q11.2 (Velocardofacial, DiGeorge) (http://www.ncbi.nlm.nih.gov/), suggesting that the phenotypic effects reported here are specific for deletion of this region of chromosome 21.
Only three of the existing reports of pure partial monosomy 21 provide confirmation by neuroimaging of the associated structural brain malformations.13,23,24 These include Estabrooks et al.,13 who first reported holoprosencephaly (HPE) detected by CT in one individual with partial monosomy 21 with deletions from 21q22.3 to the 21q telomere (qter) (MH in Fig. 3). On the basis of this and other cases, an HPE minimal critical region in 21q22.3 was defined as D21S113 to qter.14 This overlaps the consensus region for cortical dysplasia defined here by 3.2 Mb, which suggested that deletion of the HPE1 gene might also cause the defects reported here. However, the lack of signs of HPE sequence in these individuals does not support this, although deletion of the HPE1 gene may contribute. Chen et al.24 reported an individual with microcephaly, corpus callosum dysgenesis, and colpocephaly associated with deletion from an uncertain proximal breakpoint located between D21S156 and D21S53 through the telomere,24 supporting our conclusions and likely narrowing the centromeric border of the consensus region to D21S156. The telomeric border of the consensus region is narrowed by a number of reports suggesting that deletion of the telomeric region of 21q22.3 (D21S112-telomere) does not result in severe phenotypes.25,26 Falik-Borenstein et al.15 reported two individuals with partial monosomy 21 (MT and ST in Fig. 3), both of whom had deletions from COL6A2 to qter and had normal range intelligence. Therefore, we combine the data from these cases and suggest that deletion of an 8.4-Mb consensus region in 21q22.11-22.3 from KCNJ6 to COL6A2 is associated with cortical dysplasia (Fig. 3), and that one or more dosage-sensitive genes important for cortical development map within this region. The centromeric border is likely narrowed to D21S156 on the basis of Chen and colleagues' report,24 decreasing the consensus region to X Mb.
It is important to note that deletion of the consensus region KCNJ6-COL6A2 is not invariably associated with striking cortical dysplasia, similar to the variable expressivity seen with familial HPE in which obligate carriers of mutations of sonic hedgehog do not exhibit HPE.29 This variability is illustrated by the report of Ehling et al.,23 who described two unrelated individuals with deletions from the region of ETS2 to qter associated with developmental delay, one of whom had a reportedly normal MRI,23 which supports the morphogenetic variability expected with human mutations.
Genes located in the region KCNJ6-COL6A2 that are expressed in the developing cortex may contribute to the cortical dysplasia seen in these individuals. Although this region contains numerous genes whose functions and expression patterns in developing brain are unknown, we note that some, including DSCAM,30 are expressed in developing cortex. We further note that although the gene encoding DYRK1A is not within the candidate region, it maps directly proximal to the centromeric breakpoint, and its expression in the developing brain31,32 could therefore be disturbed. However, the consequences position effects on gene expression are likely to be more subtle than the effects of gene deletion. Therefore, although modified expression of DYRK1A may contribute to the brain defects reported here, it is unlikely to be solely responsible.
It is of interest to ask how these data provide clues to brain morphogenesis in DS. We note that the region deleted in case 1 closely mirrors the “DS critical region” in chromosome band 21q22.2-22.3 (Fig. 3). Duplication of this region has been shown to contribute to many of the features of DS, particularly the cognitive defects.7,9–11,33 We therefore compared the features of case 1, as well as cases 2 and 3, with those of DS (Table 1) to determine the phenotypic consequences of deletion versus duplication of this region. We found that deletion of this region does disturb the same systems as in DS, resulting in defects of the heart, eye, genitourinary system, and central nervous system structures, as well as in general growth retardation, although it does not give rise to the major visceral features of DS. Therefore, it may not be unreasonable to consider that a subset of dosage genes in 21q22 may influence the major systems involved in the pathogenesis of DS and disturb development of these systems in monosomy 21. Single-gene models such as knockout mice may help to identify the genes involved.
In conclusion, we suggest the hypothesis that one or more dosage-sensitive genes located in the region defined by KCNJ6-COL6A2 contributes to brain development, particularly the cerebral cortex. These results may be useful for understanding the genetic origins of neuroanatomic and cognitive deficits of DS and of mentally retarded persons with cortical dysplasia.
References
Raz N, Torres IJ, Briggs SD, Spencer WD et al. Selective neuroanatomic abnormalities in Down's syndrome and their cognitive correlates: evidence from MRI morphometry. Neurology 1995; 45: 356–366.
Aylward EH, Habbak R, Warren AC, Pulsifer MB et al. Cerebellar volume in adults with Down syndrome. Arch Neurol 1997; 54: 209–212.
Pinter JD, Eliez S, Schmitt JE, Capone GT et al. Neuroanatomy of Down's syndrome: a high-resolution MRI study. Am J Psychiatry 2001; 158: 1659–1665.
Hattori M, Fujiyama A, Taylor TD, Watanabe H et al. The DNA sequence of human chromosome 21. Nature 2000; 405: 311–319.
Gardiner K, Slavov D, Bechtel L, Davisson M . Annotation of human chromosome 21 for relevance to Down syndrome: gene structure and expression analysis. Genomics 2002; 79: 833–843.
Roland B, Cox DM, Hoar DI, Fowlow SB et al. A familial interstitial deletion of the long arm of chromosome 21. Clin Genet 1990; 37: 423–428.
Korenberg JR, Kawashima H, Pulst SM, Ikeuchi T et al. Molecular definition of a region of chromosome 21 that causes features of the Down syndrome phenotype. Am J Hum Genet 1990; 47: 236–246.
Korenberg JR, Kalousek DK, Anneren G, Pulst SM et al. Deletion of chromosome 21 and normal intelligence: molecular definition of the lesion. Hum Genet 1991; 87: 112–118.
Korenberg JR, Bradley C, Disteche CM . Down syndrome: molecular mapping of the congenital heart disease and duodenal stenosis. Am J Hum Genet 1992; 50: 294–302.
Chettouh Z, Croquette MF, Delobel B, Gilgenkrants S et al. Molecular mapping of 21 features associated with partial monosomy 21: involvement of the APP-SOD1 region. Am J Hum Genet 1995; 57: 62–71.
Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A et al. Chromosome 21 and Down syndrome: from genomics to pathophysiology. Nat Rev Genet 2004; 5: 725–738.
Korenberg JR, Chen XN, Schipper R, Sun Z et al. Down syndrome phenotypes: the consequences of chromosomal imbalance. Proc Natl Acad Sci U S A 1994; 91: 4997–5001.
Estabrooks LL, Rao KW, Donahue RP, Aylsworth AS . Holoprosencephaly in an infant with a minute deletion of chromosome 21(q22.3). Am J Med Genet 1990; 36: 306–309.
Muenke M, Bone LJ, Mitchell HF, Hart I et al. Physical mapping of the holoprosencephaly critical region in 21q22.3, exclusion of SIM2 as a candidate gene for holoprosencephaly, and mapping of SIM2 to a region of chromosome 21 important for Down syndrome. Am J Hum Genet 1995; 57: 1074–1079.
Falik-Borenstein TC, Pribyl TM, Pulst SM, Van Dyke DL et al. Stable ring chromosome 21: molecular and clinical definition of the lesion. Am J Med Genet 1992; 42: 22–28.
Korenberg JR, Chen XN . Human cDNA mapping using a high-resolution R-banding technique and fluorescence in situ hybridization. Cytogenet Cell Genet 1995; 69: 196–200.
Korenberg JR, Pulst SM, Neve RL, West R . The Alzheimer amyloid precursor protein maps to human chromosome 21 bands q21.105-q21.05. Genomics 1989; 5: 124–127.
Owen MJ, James LA, Hardy JA, Williamson R et al. Physical mapping around the Alzheimer disease locus on the proximal long arm of chromosome 21. Am J Hum Genet 1990; 46: 316–322.
Cox DR, Shimizu N . Report of the committee on the genetic constitution of chromosome 21. Cytogenet Cell Genet 1990; 55: 235–244.
Gardiner K, Davisson M . The sequence of human chromosome 21 and implications for research into Down syndrome. Genome Biol 2000; 1: REVIEWS0002.
Korenberg JR, Yang-Feng T, Schreck R, Chen XN . Using fluorescence in situ hybridization (FISH) in genome mapping. Trends Biotechnol 1992; 10: 27–32.
Theodoropoulos DS, Cowan JM, Elias ER, Cole C . Physical findings in 21q22 deletion suggest critical region for 21q- phenotype in q22. Am J Med Genet 1995; 59: 161–163.
Ehling D, Kennerknecht I, Junge A, Prager B et al. Mild phenotype in two unrelated patients with a partial deletion of 21q22.2-q22.3 defined by FISH and molecular studies. Am J Med Genet A 2004; 131: 265–272.
Chen CP, Lin SP, Chern SR, Lee CC et al. De novo satellited 21q associated with corpus callosum dysgenesis, colpocephaly, a concealed penis, congenital heart defects, and developmental delay. Genet Couns 2004; 15: 437–442.
McGinniss MJ, Kazazian HH Jr, Stetten G, Petersen MB et al. Mechanisms of ring chromosome formation in 11 cases of human ring chromosome 21. Am J Hum Genet 1992; 50: 15–28.
Pangalos C, Theophile D, Sinet PM, Marks A et al. No significant effect of monosomy for distal 21q22.3 on the Down syndrome phenotype in “mirror” duplications of chromosome 21. Am J Hum Genet 1992; 51: 1240–1250.
Dobyns WB, Stratton RF, Parke JT, Greenberg F et al. Miller-Dieker syndrome: lissencephaly and monosomy 17p. J Pediatr 1983; 102: 552–558.
Berry-Kravis E, Israel J . X-linked pachygyria and agenesis of the corpus callosum: evidence for an X chromosome lissencephaly locus. Ann Neurol 1994; 36: 229–233.
Muenke M, Gurrieri F, Bay C, Yi DH et al. Linkage of a human brain malformation, familial holoprosencephaly, to chromosome 7 and evidence for genetic heterogeneity. Proc Natl Acad Sci U S A 1994; 91: 8102–8106.
Barlow GM, Micales B, Chen XN, Lyons GE et al. Mammalian DSCAMs: roles in the development of the spinal cord, cortex, and cerebellum?. Biochem Biophys Res Commun 2002; 293: 881–891.
Song WJ, Sternberg LR, Kasten-Sportes C, Keuren ML et al. Isolation of human and murine homologues of the Drosophila minibrain gene: human homologue maps to 21q22.2 in the Down syndrome “critical region”. Genomics 1996; 38: 331–339.
Song WJ, Chung SH, Kurnit DM . The murine Dyrk protein maps to chromosome 16, localizes to the nucleus, and can form multimers. Biochem Biophys Res Commun 1997; 231: 640–644.
Delabar JM, Theophile D, Rahmani Z, Chettouh Z et al. Molecular mapping of twenty-four features of Down syndrome on chromosome 21. Eur J Hum Genet 1993; 1: 114–124.
Acknowledgements
This work was supported by National Institutes of Health grants HD045225 and HD17449 (J.R.K.). J.R.K. holds the Geri and Richard Brawerman Chair of Molecular Genetics. The authors also thank the individuals described in this article, together with their families, for their patience and cooperation during the course of this study.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Yao, G., Chen, XN., Flores-Sarnat, L. et al. Deletion of chromosome 21 disturbs human brain morphogenesis. Genet Med 8, 1–7 (2006). https://doi.org/10.1097/01.gim.0000195892.60506.3f
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/01.gim.0000195892.60506.3f
Keywords
This article is cited by
-
Suppression of pyramidal neuron G protein-gated inwardly rectifying K+ channel signaling impairs prelimbic cortical function and underlies stress-induced deficits in cognitive flexibility in male, but not female, mice
Neuropsychopharmacology (2021)
-
Deletion 21q22.3 and duplication 7q35q36.3 in a Colombian girl: a case report
Journal of Medical Case Reports (2016)
-
Education and Parental Involvement in Decision‐Making About Newborn Screening: Understanding Goals to Clarify Content
Journal of Genetic Counseling (2015)
-
Genomic analysis of partial 21q monosomies with variable phenotypes
European Journal of Human Genetics (2011)
