
Abstract
To identify genes and biologically relevant pathways associated with risk to develop multiple sclerosis (MS), the Genome-Wide Association Studies noise reduction method (GWAS-NR) was applied to MS genotyping data. Regions of association were defined based on the significance of linkage disequilibrium blocks. Candidate genes were cross-referenced based on a review of current literature, with attention to molecular function and directly interacting proteins. Supplementary annotations and pathway enrichment scores were generated using The Database for Annotation, Visualization and Integrated Discovery. The candidate set of 220 MS susceptibility genes prioritized by GWAS-NR was highly enriched with genes involved in biological pathways related to positive regulation of cell, lymphocyte and leukocyte activation (P=6.1E-15, 1.2E-14 and 5.0E-14, respectively). Novel gene candidates include key regulators of NF-κB signaling and CD4+ T helper type 1 (Th1) and T helper type 17 (Th17) lineages. A large subset of MS candidate genes prioritized by GWAS-NR were found to interact in a tractable pathway regulating the NF-κB-mediated induction and infiltration of pro-inflammatory Th1/Th17 T-cell lineages, and maintenance of immune tolerance by T-regulatory cells. This mechanism provides a biological context that potentially links clinical observations in MS to the underlying genetic landscape that may confer susceptibility.
Similar content being viewed by others


Introduction
Multiple sclerosis (MS) is an autoimmune disease with unknown etiology, primarily targeting the myelin sheaths of neuronal axons in the central nervous system.1 The strongest and earliest identified genetic association with MS is within the major histocompatibility complex (MHC),2 localizing to HLA-DR2 (DRB1*1501)/DQ6 (DQB1*0602).3, 4, 5 Subsequent gene mapping and genome-wide association studies (GWAS) identified additional susceptibility loci outside of the MHC, including the IL2RA and IL7RA receptors.6, 7 Since 2007, numerous additional risk variants have been identified outside of the MHC, primarily through large-scale replication, additional GWAS and meta-analysis of GWAS.5, 6, 7, 8, 9, 10, 11 A complementary identification strategy exploited targeted genotyping of susceptibility variants across several autoimmune diseases, constructing a customized ‘ImmunoChip’ to study 194 non-MHC loci having genome-wide significant association with at least one autoimmune disease. This strategy resulted in 48 new susceptibility variants, bringing the most recent number of established MS risk variants outside of the MHC to >100.12
A variety of biological pathways have been implicated in MS etiology based on gene enrichment analysis, including cell adhesion,12, 13 leukocyte activation, apoptosis, Janus kinase (JAK)-STAT signaling,14 NF-κB activation,12 and T-cell activation and proliferation.10 Further efforts may be enhanced by the identification of novel risk loci for follow-up functional analysis, examination of common functions and molecular interactions across high-probability candidate genes, and the detailed articulation of biological mechanisms that unify clinical observations in MS with the numerous genetic variants that are suggested to affect disease liability. These represented the aims of the current study.
GWAS noise reduction (GWAS-NR) is an approach to extend the power of GWAS studies to detect novel associations beyond those detected by traditional GWAS. In a genomic context, ‘noise’ represents random differences in the distribution of alleles between cases and controls that are not driven by the signal of interest: true linkage disequilibrium (LD) with a susceptibility or causative variant. GWAS-NR amplifies association signals that are locally replicated at nearby markers and across independent data subsets, attenuating the effect of random uncorrelated noise. In simulations comparing GWAS-NR with single-marker joint analysis and Fisher’s tests, the GWAS-NR algorithm has demonstrated greater power to detect true positives in a variety of disease models.15 The GWAS-NR algorithm was applied here to genome-wide data generated by the International Multiple Sclerosis Genetics Consortium (IMSGC).
Results
The most significant association region identified by GWAS-NR was a 4.4MB span covering the MHC on chromosome 6 (29 616-33 997 kb). Using significance threshold of P<1.0E-04, 191 significant LD blocks in this region were identified, separated by an average of 9 kb and no more than 332 kb. The most significant LD block (P=2.6E-55) was 1 kb upstream of HLA-DRA, which encodes one of the HLA class II alpha chain paralogues.
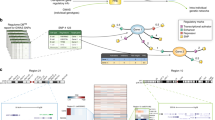
Among 162 363 LD blocks outside of the MHC, 221 significant blocks (P<1.0E-4) were identified, 215 of which had at least one other significant block in close proximity. Merging adjacent significant blocks separated by <250 kb produced 84 distinct genomic clusters comprising the strongest GWAS-NR association signals in MS outside of the MHC, corresponding to 220 candidate genes. These association clusters and candidate genes are presented in Figure 1.

Significant non-MHC association clusters in MS identified by GWAS-NR. Association clusters are defined by one or more significant (P<1.0E-4) LD blocks separated by <250 kb. Genes nearest to SNPs within each cluster, or within 25 kb of cluster boundaries are shown. Known immune-related candidates are shown in boldface. Candidates classified as demonstrating previously reported significance (Methods section) are shown in black text, with novel candidates shown in blue. † Cluster includes adjacent marginally significant LD block (P=1.06E-4). †† STAT5A, TBX21, IL12RB1, CD6, NFKBIZ and STAT1 are included due to immediate adjacency to corresponding association clusters.
Significant association clusters identified by GWAS-NR that did not overlap regions within 1 MB of the 97 high-confidence loci based on the IMSGC ImmunoChip analysis12 were examined, where the 1 MB threshold was chosen to conservatively define novel findings. Among the novel immune-related MS, susceptibility candidates within or immediately adjacent to these non-overlapping regions are: MAP3K14, which encodes NF-κB inducing kinase (NIK), a central component of non-canonical NF-κB activation; RELA, which encodes the NF-κB p65 subunit; UBASH3B, which cooperates with INPP5D to negatively regulate NF-κB signaling downstream of Fc receptor activation;16 NCOA2, a dose-dependent regulator of NF-κB activity;17 TNFAIP8, a negative regulator of NF-κB activation that maintains immune homeostasis;18 KAT5, which acts as a molecular bridge between RELA activation and NF-κB target genes;19 CSF2RB, a mediator of cytokine-induced JAK/STAT signaling cascades;20 STIM2, which regulates autoimmune effector functions of Th1 and T helper type 17 (Th17) cells;21 TXK, a Th1 cell-specific transcription factor;22 DPP4 (CD26), a T-cell activation marker implicated in autoimmune pathology and highly expressed on Th17 cells;23 CADM1, a cell adhesion molecule that promotes cell-mediated cytotoxicity;24 DAP, a positive regulator of IFN-γ mediated cell-death25 and RGCC, a regulator of T-cell-mediated apoptosis.26
Significant GWAS-NR association clusters more than 1 MB from ImmunoChip loci also include three immune-related genes for which suggestive association with MS has been reported in prior studies. These are: NFKBIZ, which encodes IkBζ, a repressor of NF-κB signaling activator upstream of IL6 production and Th17 differentiation;27, 28 INPP5D, which encodes a negative regulator of NF-κB signaling and IL12 production in macrophages;29, 30, 31 and TEC, which encodes a protein tyrosine kinase required for lipopolysaccharide-induced expression of tumor necrosis factor (TNF)-α, IL1β and IL6.14, 32
Six genes identified by GWAS-NR and having previously reported significance in MS are not covered within 1 MB of the 97 ImmunoChip loci. These include MERTK, a receptor tyrosine kinase that negatively regulates lipopolysaccharide-induced NF-κB activation;33, 34 MALT1, which forms a trimeric complex with BCL10 and CARD11 to regulate NF-κB signaling necessary for the generation of neuroinflammatory Th17 cells;35 IL12B, an activator of STAT4 upstream of Th1 differentiation;36 BATF, which encodes basic leucine zipper transcription factor, which along with RORC (RORγt) is part of the molecular signature for all IL-17 producing cells in vivo,37, 38 SOX8, a transcription factor expressed in neural crest development,11, 39 and ZNF746, which transcriptionally represses the PPAR-γ coactivator.40
To evaluate the stringency of the a priori LD block significance threshold (P<1.0E-04), LD blocks were identified corresponding to 111 genes closest (defined by genomic position) to loci having previously reported significance in MS based on single-marker analyses. Fifty-nine of these genes (53.2%) were included among LD blocks meeting this significance threshold, with four additional genes of previously reported significance captured by clusters joining significant LD blocks within 250 kb proximity. An alternative LD block significance level of 1.0E-03 would increase the number of susceptibility candidates from 220 to 568.
Based on 162 363 LD blocks outside of the MHC, 30 blocks satisfying a Bonferroni-corrected significance level of P<3.1E-07 were identified. Known immune-related candidates in these LD blocks, based on ImmPort classification (https://immport.niaid.nih.gov) and review of current literature, included CLEC16A, CD86 (B7-2), PTGER4, IL2RA, FAM213B, GFI1, TNFRSF1A, EOMES, STAT3, FCRL3, RPS6KB1, GPR65, TNFSF14 and CD58. Although MANBA is not classified as immune-related, the most significant LD block associated with this gene is immediately adjacent to NFKB1. Only 13 of the 111 genes with previously reported significance in MS were associated with LD blocks meeting the Bonferroni threshold.
The 220 non-MHC candidate susceptibility genes identified by GWAS-NR are listed in Supplementary Table 1. Among these candidate genes, 57 were located >1MB from significant ImmunoChip loci, with the remaining 163 candidates located within 1MB of those loci. Genes of previously reported significance in MS comprised 63 of the 220 candidates.
The 220 genes in the GWAS-NR candidate set were examined for functional enrichment in biological pathways. The strongest functional enrichment reported by DAVID41, 42 for the candidate gene set was positive regulation of cell activation (GO term 0050867, P=6.1E-15) with a number of highly synonymous pathways (for example, positive regulation of lymphocyte activation, positive regulation of leukocyte activation) reflecting the same subset of genes. Additional significant pathways included positive regulation of immune system process (GO term 0002684, P=2.6E-13), regulation of lymphocyte proliferation (GO term 00050670, P=2.2E-09), regulation of lymphocyte mediated immunity (GO term 0002706, P=8.2E-09) and regulation of cytokine production (GO term 0001817, P=1.7E-08). Common biological functions of MS candidate gene set identified by GWAS-NR, and specific genes responsible for pathway enrichment, are reported in Supplementary Table 2.
For comparative purposes, genes outside the MHC were ranked according their smallest single-marker joint analysis P-value. The single-marker significance threshold required to capture 220 candidate genes was 5.4E-06. The strongest functional enrichment reported by DAVID for this gene set was also positive regulation of cell activation, but with lower significance (P=2.3E-12) compared with the GWAS-NR candidate set, resulting from the inclusion of fewer relevant genes in this pathway. Compared with the significance of pathway enrichment among GWAS-NR candidates, uniformly less significant enrichment was observed among the 10 highest-ranked biological pathways using these genes defined by single-marker significance, and in 90% of the 20 highest-ranked pathways, even when matched for identical Gene Ontology terms.
To identify potentially important candidates not captured by GWAS-NR at the a priori significance level of 1.0E-04, regions within 250 kb of the 97 ImmunoChip loci were examined. 511 genes in these regions were not included in the candidate set identified by GWAS-NR. As observed among the GWAS-NR candidate set, the strongest functional enrichments reported by DAVID for these additional genes were in domains related to cell-mediated immunity, particularly regulation of lymphocyte differentiation (GO term 00045619, P=1.8E-04) and regulation of leukocyte activation (GO term 0002694, P=3.1E-04).
Discussion
Our results indicate that outside of the MHC region, the genetic background of MS is enriched with numerous variants that regulate the activation and proliferation of immune effector cells, extending genetic evidence that implicates MS as an autoimmune inflammatory condition.6 GWAS-NR prioritizes a combined 5.2 MB span of the non-MHC genome, including several novel regions of association. The resulting candidate gene set includes central components of NF-κB signaling (MAP3K14, RELA), and provides novel or additional evidence for regulation of NF-κB activation by membrane-bound receptors (UBASH3B, INPP5D, NCOA2, TNFAIP8, KAT5 and NFKBIZ). These results reinforce a previously suggestive role for NF-κB activation in MS implicated by significant association loci adjacent to BCL10 and CARD11 (ref. 12) and functional analysis of MS-associated variants proximal to NF-κB1 and in an intron of TNFRSF1A.43 Additional novel candidates identified by GWAS-NR include regulators of CD4+ Th1 and Th17 induction (STIM2, DPP4, TXK), and apoptosis (CADM1, DAP, RGCC), providing further support for involvement of these pathways in MS.10, 14 Several immune-related candidates identified by GWAS-NR within 1 MB of significant ImmunoChip loci are not included among genes with previously reported significance in MS, including SOCS1, TNFRSF14, GFI1, STAT1, STAT5A, CD5, KLRB1, AMICA1, IL12RB1, TBKBP1, CD80 and REL.
Examination of LD block scores of genes with previously reported significance in MS suggests that the a priori LD block significance level (P<1.0E-04) chosen for reporting purposes is conservative. The identified blocks span ~0.2% of the non-MHC genome, and capture over 50% of the genes previously reported as proximal to at least one genome-wide significant single-nucleotide polymorphism (SNP) in MS based on previous published single-marker analyses.
Although the GWAS-NR algorithm is well-suited to identifying and prioritizing candidates and regions for follow-up study, the filtering process results in P-values that are less likely to achieve genome-wide significance, compared with single-marker P-values that may benefit from contributions of noise. Moreover, GWAS-NR can achieve no advantage over single-marker analysis when flanking markers provide little supplementary information, which may occur when a true locus is typed directly and a single-marker association method is used.15 It is likely that additional genetic variation related to MS remains unidentified by any genome-wide association technique. Thus, the candidate susceptibility loci identified by GWAS-NR are not exhaustive, and can be viewed as complementary to high-confidence loci identified by alternative methods.
GWAS-NR reduces statistical noise by exploiting local correlation of association signals across multiple data sets, whereas biological pathway analysis exploits the concurrence of multiple candidate genes within various biological processes to identify functions that are highly represented among these candidate genes. The grouping of candidates into local association clusters can also provide informative context, both because association signals at a given locus may reflect a causal variant at a different location in LD, and because local clusters of susceptibility loci may include functionally related genes. All of these approaches may be viewed as noise reduction strategies. Applied jointly, we observe that the majority of association clusters identified by GWAS-NR include one or more immune-related genes having molecular functions common to those in other clusters. Supplementary Table 1 includes additional references related to individual candidates.
Although it is not possible to exclude a role in MS for candidate susceptibility genes that are uncharacterized or have no common or known immunological function, numerous immune-related candidates identified and prioritized by GWAS-NR have well-established roles in biologically relevant pathways such as TNF-α signaling, NF-κB transcription, cytokine production, JAK/STAT signaling, T-cell differentiation and cell adhesion. Several of these biological functions have been individually implicated in MS based on gene previous enrichment analyses.10, 12, 13, 14, 44
We propose that in MS, these functions are not distinct from one another, but instead comprise a coordinated mechanism that may contribute to MS pathology. Based on previously reported molecular functions and interactions among the MS candidate genes identified by GWAS-NR, the genes responsible for significant enrichment in common biological functions were found to interact in a tractable pathway regulating the NF-κB-mediated induction and infiltration of pro-inflammatory Th1/Th17 T-cell lineages, and the maintenance of immune tolerance by suppressive T-regulatory cells. Figure 2, constructed on the basis of reported interactions among the MS susceptibility candidate genes prioritized by GWAS-NR, provides a graphic overview of this pathway.

NF-κB-mediated regulation of inflammatory and immunosuppressive CD4+ T-cell lineages. Proposed mechanism of MS pathology implicated by pathway analysis of GWAS-NR candidate genes. This simplified schematic presents an abridged set of interactions in an exploded format for tractability, uses generic labels for several homologous candidates (for example, B7 represents both CD80 and CD86), and does not individually depict each candidate gene implicated in current literature as having relevance to this pathway. Activation of membrane-bound T-cell receptors (TCR),75 pattern recognition receptors such as dendritic-cell Toll-like receptors and C-type lectins76, 77, 78 and receptors of the TNF superfamily79, 80 initiate signal transduction cascades leading to the activation of NF-κB transcriptional programs through canonical and non-canonical pathways, resulting in cytokine production that determines the balance between inflammatory Th1 and Th17 cells, and immunosuppressive regulatory T cells via JAK/STAT signaling to lineage-specific transcription factors.81 NF-κB activation also promotes the expression of cell adhesion molecules on vascular endothelia, which enable the infiltration of inflammatory T cells across the blood–brain barrier.62, 82 CLEC, C-type lectin; FcR, Fc receptor; LPS, lipopolysaccharide; TLR, Toll-like receptor; TNFR, TNF-α receptor; Th1, T helper type 1 cell; Treg, T-regulatory cell.
We propose this mechanism as relevant to MS pathology based on numerous risk genes implicated by GWAS-NR that act as key mediators with cooperative roles at each level of this pathway, comprising both novel candidates and previously reported MS risk genes. The regulation of NF-κB-mediated Th1/Th17 inflammation and Treg tolerance involves a chain of interactions comprising receptor activation, signal transduction, NF-κB transcription, cytokine production, JAK/STAT signaling, T-cell induction and adhesion-mediated cell motility, and can be well-described with specific reference to interactions between these candidate genes.
Several TNF family genes are included in the GWAS-NR candidate set. TNF family members are among the best-characterized inducers of NF-κB signaling.45 NF-κB signaling is a critical regulator of the balance between inflammation and tolerance, as NF-κB activation appears to be required both for the induction of pro-inflammatory Th17 cells and immunosuppressive Treg cells.46
Activation of tumor necrosis superfamily member TNFRSF14 by its ligand TNFSF14, both candidates identified by GWAS-NR, acts as a T-cell costimulatory signal that results in NF-κB-mediated induction of pro-inflammatory genes.47 Induction of NF-κB by TNF-α also promotes the expression of cell adhesion molecules ICAM1 and VCAM1.48 Although nonspecific anti-TNF therapy does not result in remission of symptoms in MS, selective blocking of TNFRSF1A significantly suppresses the infiltration of inflammatory Th1 and Th17 cells, and significantly improves clinical outcomes in murine experimental autoimmune encelphalomyelitis, a primary animal model of MS pathology.49 Conversely, defective clearance of TNFRSF1A accumulation is associated with NF-κB activation, excessive IL1β secretion and chronic inflammation.50
NF-κB is a transcription factor composed of homo- or heterodimers of five subunits: RELA, RELB, REL, NFKB1 and NFKB2. Canonical NF-κB activation triggers the production of cytokines such as IL1β, IL651 and IL12, and is inhibited by RPS6KB1 (S6K1). Knockdown of this inhibition results in enhanced production of inflammatory cytokines.19 Cytokine response to NF-κB signaling is dependent on the profile of subunit activation,52 and is subject to coordinated regulation by other signaling pathways.
Optimal T-cell receptor activation of the canonical NF-κB pathway requires costimulation53 by CD80 (B7-1) or CD86 (B7-2), and is mediated by a trimeric complex of adapter proteins known as the CARD11-BCL10-MALT1 (CBM) complex. The non-canonical NF-κB pathway involves activation of MAP3K14 (‘NIK’) by members of the TNF family, such as by CD40 activation of TRAF3. All of the foregoing are among MS risk candidates identified by GWAS-NR. Expression of NIK by dendritic cells is necessary to establish Th1 and Th17 responses.54 NIK also coordinates signaling by T-cell receptor and IL6 to activate STAT3, contributing to the induction of Th17 cells.55
CD4+ T-cells differentiate into effector cells or T-regulatory cells depending on lineage-specific signals determined by the pathogen and resulting microenvironment that is encountered. Cytokine production activates the JAK – signal transducers and activators of transcription (STAT) pathway. Uninfected dendritic cells produce TGFβ, which activates STAT5 induction of FOXP3, the lineage-specific transcription factor for regulatory T cells. Th1 differentiation is dependent on IL12 and IFN-γ activation of STAT4 and STAT1, respectively, resulting in induction of the transcription factor TBX21. The transcription factor EOMES can compensate for TBX21 deficiency and favors Th1 development.56 The Th17 differentiation program involves IL6 activation of STAT3, which is enhanced by SOCS1, and induces the lineage-specific transcription factor RORC.57 As IL6 is one of the most strongly induced NF-κB-dependent cytokines,58 NF-κB has a particularly important role in the induction of inflammatory Th17 cells. REL and RELA directly induce RORC, and NF-κB also regulates the production of IL6 and IL23.18
The cytokines IL2 and IL7 help to regulate the balance between inflammation and tolerance. Expression of IL2RA and the presence of IL2 are critical for the generation of Treg cells.59 IL2 has a central role in the maintenance of peripheral tolerance by eliminating self-reactive T cells, and knockout of IL2RA (CD25) results in autoimmune disease in mice.60 IL7 is essential for T-cell survival, and the high-affinity receptor IL7R is also expressed on Treg cells. However, the presence of IL7 impairs the ability of Treg cells to suppress proliferation of autoreactive T cells in response to T-cell receptor activation by autoantigens, promoting their uncontrolled expansion IL7-rich environments.61
Inflammatory T cells do not normally cross the blood–brain barrier unless it is disrupted. The expression of cell adhesion molecules ICAM1 and VCAM1 on vascular endothelia is promoted by NF-κB activation and inhibited by S1PR5. Knockdown of S1PR5 abolishes quiescence of the blood–brain barrier and increases the expression of ICAM1 and VCAM1 in an NF-κB-dependent manner,62 creating a permissive environment for the infiltration of inflammatory T cells into the CNS. AMICA1 also contributes to the binding of leukocytes expressing α4β1 integrin (VLA-4) on their plasma membranes to endothelial adhesion molecules.63
The regulation of NF-κB-mediated Th1/Th17 inflammation and Treg tolerance implicated by the GWAS-NR candidate gene set is consistent with clinical observations in MS. Activated T cells from patients with MS produce elevated levels of IL-17. Blockade of IL6R signaling reduces this production, elevates IL10 release by activated CD4+ T-cells and restores the ability of hydrocortisone to inhibit T-cell proliferation.64 A positive association between Th17 and Treg cell percentages is found in MS patients with remission, but not during relapse,65 while prevalence of Th17 cells in CSF is significantly elevated during relapse.66 Elevated expression of IFN-γ is observed during both early phase and relapse,67 and STAT3 expression is persistently higher in CD4+ T-cells in patients who subsequently convert to clinically defined MS than in patients who do not and controls.67 This cytokine profile is consistent with infiltration of pro-inflammatory Th1/Th17 lineages during exacerbations, with immunosuppression by Treg cells during remission.
At the transcriptional level, analysis of the T-cell transcriptome identifies aberrant regulation of gene expression by NF-κB as a biomarker of relapse in MS based on 43 differentially expressed genes.68 Upregulation of nuclear staining for both NF-κB and JNK was found on a large proportion of oligodendrocytes at the edge of active lesions and on microglia/macrophages throughout actively demyelenating plaques.69
Materials and methods
The data set used for analysis included 9772 individuals with MS and 17 376 healthy controls from 15 countries; the majority of which were recruited by the IMSGC and the Wellcome Trust Case Control Consortium. All individuals in this study gave informed consent with approval from the relevant local Ethical Committees or Institutional Review Boards. Details of the ascertainment scheme and quality control of samples were described previously.10 To maximize the capability of GWAS-NR in amplifying association signals that are locally replicated across loci in independent data subsets, the samples were divided into seven population specific stratum: Australia (793 MS, 2160 control), Central Europe (2242 MS, 2046 control), Mediterranean (950 MS, 1317 control), Finland (581 MS, 2165 control), Scandinavia (1970 MS, 2049 control), United Kingdom (1854 MS, 5175 control) and United States (1382 MS, 2464 control).
Therefore, in addition to the SNP quality control that was done previously,10 resulting in 465 434 autosomal SNPs, quality control was done within each of these assigned strata. This included removing SNPs with low call rate (<95%), with differential missingness between individuals with MS and healthy controls (P<1.0E-03), and which were out of Hardy-Weinberg equilibrium (HWE) (P<1.0E-05). This resulted in 447 419 SNPs which passed quality control in all seven strata and were subsequently analyzed.
Eigenstrat70 was run separately within each stratum using 43K independent SNPs (R2<0.1) with minor allele frequency ⩾0.1 across the genome. Logistic regression analysis was run within each stratum using PLINK71 and adjusting for the first five principal components from Eigenstrat to account for residual population stratification. After sample size adjustment, the genomic inflation factor in each stratum was <1.05, indicating minimal evidence for residual population stratification.72
A joint P-value across all strata was obtained using an inverse-variance meta-analysis of the seven strata, under a fixed effects model. The P-values from each of the strata and from the joint analysis were then input into the GWAS-NR algorithm. The GWAS-NR algorithm (version 2) was executed as a Matlab script and is available from http://hihg.med.miami.edu/software-download/gwas-nr-version-2.0. Noise reduction is achieved by applying a linear filter within a sliding window to identify genomic regions demonstrating correlated profiles of association across multiple subsets; capturing association signals from surrounding markers in LD and suppressing signals that are not replicated in multiple data sets or across flanking markers. The algorithm produces a weighted P-value for each SNP, for use in prioritizing genomic regions and associated genes for follow-up study.15
The GWAS-NR and joint analysis methods were compared for sensitivity and specificity in identifying target loci defined, for comparative purposes, as all genotyped loci within 250 kb of 97 independent MS association signals previously identified by the IMSGC using a custom ‘ImmunoChip’ genotyping array with 29 300 subjects with MS and 50 794 controls.12 Area under the receiver operating characteristic curve was 0.635 for joint analysis, and 0.719 for GWAS-NR. At each level of specificity, GWAS-NR demonstrated higher sensitivity than joint analysis in identifying these target loci.
To evaluate the results of GWAS-NR from multiple perspectives, significant loci were examined across individual genes, LD blocks, and association clusters defined by multiple significant LD blocks in close proximity. This context was viewed as potentially informative as strong association signals at a given marker may reflect a causal variant at a different location in LD with that marker, and because physical clusters of susceptibility loci in the same region of association may include functionally related genes.73
LD blocks were defined using PLINK71 and according to the method proposed by Gabriel et al.74 Single SNPs not in an LD block with surrounding markers and between two LD blocks were treated as single-SNP blocks. Any SNP located between two SNPs within the same LD block was assigned the same block ID.
The significance of these LD blocks was computed using the Truncated Product Method of Zaykin.74 A combined score for the markers in each LD block was calculated, based on the product of GWAS-NR P-values that were below a threshold of 0.05. A Monte Carlo algorithm was used to test the significance of the combined score. To generate a reduced set of high-priority association candidates, an a priori significance level of P<1.0E-04 was chosen for reporting purposes.
To identify distinct genomic regions comprising the strongest GWAS-NR association signals, association clusters were defined by joining adjacent significant LD blocks (P<1.0E-04) separated by <250 kb. For clusters comprising more than one LD block, the lowest P-value among these blocks was identified for purposes of sorting and discussion. The candidate gene set was defined to include the gene nearest (defined by genomic position) to each SNP within each cluster, as well as the gene nearest to each SNP within 25 kb of cluster boundaries. Immune-related genes and marginally significant LD blocks (P<1.1E-04) immediately adjacent to significant association clusters were also examined as potential candidates.
Candidate genes were classified as demonstrating previously reported significance in MS if included among 111 genes reported by the IMSGC as proximal to a SNP demonstrating genome-wide significance (P<5.0E-08) or strong association (P<5.0E-07) based on GWAS10, 11 and ImmunoChip genotyping analysis.12 Although this classification is indicative of candidate genes corresponding to statistically significant and previously reported association loci, it does not represent an exhaustive list of genes that may otherwise be implicated in MS, and the associated variants may not be established as relevant to MS based on functional evidence.
To investigate functional relationships among genes in the candidate set, each candidate was manually annotated and cross-referenced, based on a review of current literature, with attention to molecular function and directly interacting proteins. Candidate genes were classified as immune-related based on reported involvement in immune function, or inclusion in the Comprehensive List of Immune-Related Genes maintained by The Immunology Database and Analysis Portal (ImmPort https://immport.niaid.nih.gov). Supplementary functional annotations and pathway enrichment scores were generated using DAVID (The Database for Annotation, Visualization and Integrated Discovery) version 6.7.41, 42
Conclusions
The GWAS-NR was applied to MS data, with the objective of prioritizing genomic regions and associated genes for biological pathway analysis, targeted sequencing and follow-up functional analysis. GWAS-NR prioritizes a combined 5.2 MB span comprising ~0.2% of the non-MHC genome. The resulting candidate set of 220 MS susceptibility genes is highly enriched with genes involved in the positive regulation of cell-mediated immunity. Novel candidates include key regulators of NF-κB signaling and regulation of CD4+Th1 and Th17 lineages. Based on previously reported molecular functions and interactions, a large subset of the MS candidate genes were found to interact in a tractable pathway regulating the NF-κB-mediated induction and infiltration of pro-inflammatory Th1/Th17 T-cell lineages, and the maintenance of immune tolerance by suppressive T-regulatory cells. This mechanism provides a biological context that potentially links clinical observations in MS to the underlying genetic landscape that may confer susceptibility, and may help to inform efforts to develop targeted therapies.
References
DeLuca J, Nocentini U . Neuropsychological, medical and rehabilitative management of persons with multiple sclerosis. NeuroRehabilitation 2011; 29: 197–219.
Sawcer S, Jones HB, Feakes R, Gray J, Smaldon N, Chataway J et al. A genome screen in multiple sclerosis reveals susceptibility loci on chromosome 6p21 and 17q22. Nat Genet 1996; 13: 464–476.
Olerup O, Hillert J . HLA class II-associated genetic susceptibility in multiple sclerosis: a critical evaluation. Tissue Antigens 1991; 38: 1–15.
Barcellos LF, Sawcer S, Ramsay PP, Baranzini SE, Thomson G, Briggs F et al. Heterogeneity at the HLA-DRB1 locus and risk for multiple sclerosis. Hum Mol Genet 2006; 15: 2813–2824.
Patsopoulos NA et althe Bayer Pharma MS Genetics Working Group, the Steering Committees of Studies Evaluating IFNbeta-1b and a CCR1-Antagonist, ANZgene Consortium, GeneMSA, International Multiple Sclerosis Genetics Consortium. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann Neurol 2011; 70: 897–912.
International Multiple Sclerosis Genetics Consortium, Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 2007; 357: 851–862.
Gregory SG, Schmidt S, Seth P, Oksenberg JR, Hart J, Prokop A et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet 2007; 39: 1083–1091.
International Multiple Sclerosis Genetics Consortium (IMSGC). Comprehensive follow-up of the first genome-wide association study of multiple sclerosis identifies KIF21B and TMEM39A as susceptibility loci. Hum Mol Genet 2010; 19: 953–962.
De Jager PL, Jia X, Wang J, de Bakker PI, Ottoboni L, Aggarwal NT et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet 2009; 41: 776–782.
International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2, Sawcer S, Hellenthal G, Pirinen M, Spencer CC et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011; 476: 214–219.
International Multiple Sclerosis Genetics Consortium, Lill CM, Schjeide BM, Graetz C, Ban M, Alcina A et al. MANBA, CXCR5, SOX8, RPS6KB1 and ZBTB46 are genetic risk loci for multiple sclerosis. Brain 2013; 136: 1778–1782.
International Multiple Sclerosis Genetics Consortium (IMSGC), Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 2013; 45: 1353–1360.
Damotte V, Guillot-Noel L, Patsopoulos NA, Madireddy L, El Behi M et alInternational Multiple Sclerosis Genetics Consortium. A gene pathway analysis highlights the role of cellular adhesion molecules in multiple sclerosis susceptibility. Genes Immun 2014; 15: 126–132.
International Multiple Sclerosis Genetics Consortium. Network-based multiple sclerosis pathway analysis with GWAS data from 15, 000 cases and 30,000 controls. Am J Hum Genet 2013; 92: 854–865.
Hussman JP, Chung RH, Griswold AJ, Jaworkski JM, Salyakina D, Ma D et al. A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Mol Autism 2011; 2: 1.
de Castro RO, Zhang J, Groves JR, Barbu EA, Siraganian RP . Once phosphorylated, tyrosines in carboxyl terminus of protein-tyrosine kinase Syk interact with signaling proteins, including TULA-2, a negative regulator of mast cell degranulation. J Biol Chem 2012; 287: 8194–8204.
Nojek IM, Werbajh SE, Colo GP, Rubio FM, Franco LD, Nahmod VE et al. Different enzymatic activities recruitment by specific domains of TIF2 are involved in NF-kappaB transactivation. Medicina (B Aires) 2004; 64: 135–138.
Sun SC, Chang JH, Jin J . Regulation of nuclear factor-kappaB in autoimmunity. Trends Immunol 2013; 34: 282–289.
Kim SY, Baik KH, Baek KH, Chah KH, Kim KA, Moon G et al. S6K1 negatively regulates TAK1 activity in the toll-like receptor signaling pathway. Mol Cell Biol 2014; 34: 510–521.
Ebner K, Bandion A, Binder BR, de Martin R, Schmid JA . GMCSF activates NF-kappaB via direct interaction of the GMCSF receptor with IkappaB kinase beta. Blood 2003; 102: 192–199.
Ma J, McCarl CA, Khalil S, Luthy K, Feske S . T-cell-specific deletion of STIM1 and STIM2 protects mice from EAE by impairing the effector functions of Th1 and Th17 cells. Eur J Immunol 2010; 40: 3028–3042.
Takeba Y, Nagafuchi H, Takeno M, Kashiwakura J, Suzuki N . Txk, a member of nonreceptor tyrosine kinase of Tec family, acts as a Th1 cell-specific transcription factor and regulates IFN-gamma gene transcription. J Immunol 2002; 168: 2365–2370.
Bengsch B, Seigel B, Flecken T, Wolanski J, Blum HE, Thimme R . Human Th17 cells express high levels of enzymatically active dipeptidylpeptidase IV (CD26). J Immunol 2012; 188: 5438–5447.
Boles KS, Barchet W, Diacovo T, Cella M, Colonna M . The tumor suppressor TSLC1/NECL-2 triggers NK-cell and CD8+ T-cell responses through the cell-surface receptor CRTAM. Blood 2005; 106: 779–786.
Feinstein E, Druck T, Kastury K, Berissi H, Goodart SA, Overhauser J et al. Assignment of DAP1 and DAPK—genes that positively mediate programmed cell death triggered by IFN-gamma—to chromosome regions 5p12.2 and 9q34.1, respectively. Genomics 1995; 29: 305–307.
Tegla CA, Cudrici CD, Azimzadeh P, Singh AK, Trippe R 3rd, Khan A et al. Dual role of Response gene to complement-32 in multiple sclerosis. Exp Mol Pathol 2013; 94: 17–28.
Okamoto K, Iwai Y, Oh-Hora M, Yamamoto M, Morio T, Aoki K et al. IkappaBzeta regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature 2010; 464: 1381–1385.
Disanto G, Kjetil Sandve G, Ricigliano VA, Pakpoor J, Berlanga-Taylor AJ, Handel AE et al. DNase hypersensitive sites and association with multiple sclerosis. Hum Mol Genet 2014; 23: 942–948.
Conde C, Rambout X, Lebrun M, Lecat A, Di Valentin E, Dequiedt F et al. The inositol phosphatase SHIP-1 inhibits NOD2-induced NF-kappaB activation by disturbing the interaction of XIAP with RIP2. PLoS One 2012; 7: e41005.
Mahurkar S, Moldovan M, Suppiah V, O'Doherty C . Identification of shared genes and pathways: a comparative study of multiple sclerosis susceptibility, severity and response to interferon beta treatment. PLoS One 2013; 8: e57655.
Hadidi S, Antignano F, Hughes MR, Wang SK, Snyder K, Sammis GM et al. Myeloid cell-specific expression of Ship1 regulates IL-12 production and immunity to helminth infection. Mucosal Immunol 2012; 5: 535–543.
Zemans RL, Arndt PG . Tec kinases regulate actin assembly and cytokine expression in LPS-stimulated human neutrophils via JNK activation. Cell Immunol 2009; 258: 90–97.
Lee YJ, Han JY, Byun J, Park HJ, Park EM, Chong YH et al. Inhibiting Mer receptor tyrosine kinase suppresses STAT1, SOCS1/3, and NF-kappaB activation and enhances inflammatory responses in lipopolysaccharide-induced acute lung injury. J Leukoc Biol 2012; 91: 921–932.
Ma GZ, Stankovich J Australia and New Zealand Multiple Sclerosis Genetics Consortium (ANZgene), Kilpatrick TJ, Binder MD, Field J . Polymorphisms in the receptor tyrosine kinase MERTK gene are associated with multiple sclerosis susceptibility. PLoS One 2011; 6: e16964.
Brustle A, Brenner D, Knobbe CB, Lang PA, Virtanen C, Hershenfield BM et al. The NF-kappaB regulator MALT1 determines the encephalitogenic potential of Th17 cells. J Clin Invest 2012; 122: 4698–4709.
Varade J, Comabella M, Ortiz MA, Arroyo R, Fernandez O, Pinto-Medel MJ et al. Replication study of 10 genes showing evidence for association with multiple sclerosis: validation of TMEM39A, IL12B and CBLB [correction of CLBL] genes. Mult Scler 2012; 18: 959–965.
Jordan-Williams KL, Poston S, Taparowsky EJ . BATF regulates the development and function of IL-17 producing iNKT cells. BMC Immunol 2013; 14: 16.
Akkad DA, Bellenberg B, Esser S, Weiler F, Epplen JT, Gold R et al. Multiple sclerosis risk loci correlate with cervical cord atrophy and may explain the course of disability. Neurogenetics 2015; 16: 161–168.
O'Donnell M, Hong CS, Huang X, Delnicki RJ, Saint-Jeannet JP . Functional analysis of Sox8 during neural crest development in Xenopus. Development 2006; 133: 3817–3826.
Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O et al. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell 2011; 144: 689–702.
Huang da W, Sherman BT, Lempicki RA . Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57.
Huang da W, Sherman BT, Lempicki RA . Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37: 1–13.
Housley W, Fernandez S, Vera K, Murikinati S, Grutzendler J, Cuerdon N et al. Genetic variants associated with autoimmunity drive NFκB signaling and responses to inflammatory stimuli. Sci Transl Med 2015; 7: 291ra93.
Baranzini SE, Galwey NW, Wang J, Khankhanian P, Lindberg R, Pelletier D et al. Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum Mol Genet 2009; 18: 2078–2090.
Hayden MS, Ghosh S . Regulation of NF-kappaB by TNF family cytokines. Semin Immunol 2014; 26: 253–266.
Ruan Q, Chen YH . Nuclear factor-kappaB in immunity and inflammation: the Treg and Th17 connection. Adv Exp Med Biol 2012; 946: 207–221.
Cheung TC, Steinberg MW, Oborne LM, Macauley MG, Fukuyama S, Sanjo H et al. Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proc Natl Acad Sci USA 2009; 106: 6244–6249.
Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T . Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J 1995; 9: 899–909.
Nomura T, Abe Y, Kamada H, Shibata H, Kayamuro H, Inoue M et al. Therapeutic effect of PEGylated TNFR1-selective antagonistic mutant TNF in experimental autoimmune encephalomyelitis mice. J Control Release 2011; 149: 8–14.
Bachetti T, Chiesa S, Castagnola P, Bani D, Di Zanni E, Omenetti A et al. Autophagy contributes to inflammation in patients with TNFR-associated periodic syndrome (TRAPS). Ann Rheum Dis 2013; 72: 1044–1052.
Young AM, Campbell EC, Lynch S, Dunn MH, Powis SJ, Suckling J . Regional susceptibility to TNF-alpha induction of murine brain inflammation via classical IKK/NF-kappaB signalling. PLoS One 2012; 7: e39049.
Gu L, Ning H, Qian X, Huang Q, Hou R, Almourani R et al. Suppression of IL-12 production by tristetraprolin through blocking NF-kcyB nuclear translocation. J Immunol 2013; 191: 3922–3930.
Schmitz ML, Krappmann D . Controlling NF-kappaB activation in T cells by costimulatory receptors. Cell Death Differ 2006; 13: 834–842.
Hofmann J, Mair F, Greter M, Schmidt-Supprian M, Becher B . NIK signaling in dendritic cells but not in T cells is required for the development of effector T cells and cell-mediated immune responses. J Exp Med 2011; 208: 1917–1929.
Jin W, Zhou XF, Yu J, Cheng X, Sun SC . Regulation of Th17 cell differentiation and EAE induction by MAP3K NIK. Blood 2009; 113: 6603–6610.
Yang Y, Xu J, Niu Y, Bromberg JS, Ding Y . T-bet and eomesodermin play critical roles in directing T cell differentiation to Th1 versus Th17. J Immunol 2008; 181: 8700–8710.
Yoshimura A, Suzuki M, Sakaguchi R, Hanada T, Yasukawa H . SOCS, inflammation, and autoimmunity. Front Immunol 2012; 3: 20.
Brasier AR . The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc Res 2010; 86: 211–218.
Shalev I, Schmelzle M, Robson SC, Levy G . Making sense of regulatory T cell suppressive function. Semin Immunol 2011; 23: 282–292.
Waldmann TA, Dubois S, Tagaya Y . Contrasting roles of IL-2 and IL-15 in the life and death of lymphocytes: implications for immunotherapy. Immunity 2001; 14: 105–110.
Heninger AK, Theil A, Wilhelm C, Petzold C, Huebel N, Kretschmer K et al. IL-7 abrogates suppressive activity of human CD4+CD25+FOXP3+ regulatory T cells and allows expansion of alloreactive and autoreactive T cells. J Immunol 2012; 189: 5649–5658.
van Doorn R, Lopes Pinheiro MA, Kooij G, Lakeman K, van het Hof B, van der Pol SM et al. Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J Neuroinflammation 2012; 9: 133–2094-9-133.
Luissint AC, Lutz PG, Calderwood DA, Couraud PO, Bourdoulous S . JAM-L-mediated leukocyte adhesion to endothelial cells is regulated in cis by alpha4beta1 integrin activation. J Cell Biol 2008; 183: 1159–1173.
Ferreira TB, Hygino J, Barros PO, Teixeira B, Kasahara TM, Linhares UC et al. Endogenous interleukin-6 amplifies interleukin-17 production and corticoid-resistance in peripheral T cells from patients with multiple sclerosis. Immunology 2014; 143: 560–568.
Peelen E, Damoiseaux J, Smolders J, Knippenberg S, Menheere P, Tervaert JW et al. Th17 expansion in MS patients is counterbalanced by an expanded CD39+ regulatory T cell population during remission but not during relapse. J Neuroimmunol 2011; 240-241: 97–103.
Brucklacher-Waldert V, Stuerner K, Kolster M, Wolthausen J, Tolosa E . Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 2009; 132: 3329–3341.
Frisullo G, Nociti V, Iorio R, Patanella AK, Marti A, Caggiula M et al. IL17 and IFNgamma production by peripheral blood mononuclear cells from clinically isolated syndrome to secondary progressive multiple sclerosis. Cytokine 2008; 44: 22–25.
Satoh J, Misawa T, Tabunoki H, Yamamura T . Molecular network analysis of T-cell transcriptome suggests aberrant regulation of gene expression by NF-kappaB as a biomarker for relapse of multiple sclerosis. Dis Markers 2008; 25: 27–35.
Bonetti B, Stegagno C, Cannella B, Rizzuto N, Moretto G, Raine CS . Activation of NF-kappaB and c-jun transcription factors in multiple sclerosis lesions. Implications for oligodendrocyte pathology. Am J Pathol 1999; 155: 1433–1438.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D . Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38: 904–909.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
de Bakker PI, Ferreira MA, Jia X, Neale BM, Raychaudhuri S, Voight BF . Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet 2008; 17: R122–R128.
Preuss C, Riemenschneider M, Wiedmann D, Stoll M . Evolutionary dynamics of co-segregating gene clusters associated with complex diseases. PLoS One 2012; 7: e36205.
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B et al. The structure of haplotype blocks in the human genome. Science 2002; 296: 2225–2229.
Gerondakis S, Fulford TS, Messina NL, Grumont RJ . NF-kappaB control of T cell development. Nat Immunol 2014; 15: 15–25.
Brown J, Wang H, Hajishengallis GN, Martin M . TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dent Res 2011; 90: 417–427.
Wevers BA, Kaptein TM, Zijlstra-Willems EM, Theelen B, Boekhout T, Geijtenbeek TB et al. Fungal engagement of the C-type lectin mincle suppresses dectin-1-induced antifungal immunity. Cell Host Microbe 2014; 15: 494–505.
Hara H, Iizasa E, Nakaya M, Yoshida H . L-CBM signaling in lymphocyte development and function. J Blood Med 2010; 1: 93–104.
Pelzer C, Thome M . IKKα takes control of canonical NF-κB activation. Nat Immunol 2011; 12: 815–816.
Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol 2008; 9: 1364–1370.
Oh H, Ghosh S . NF-kappaB: roles and regulation in different CD4(+) T-cell subsets. Immunol Rev 2013; 252: 41–51.
Petrasch S . Follicular dendritic cells in malignant lymphomas. Curr Top Microbiol Immunol 1995; 201: 189–203.
Acknowledgements
We thank the International Multiple Sclerosis Genetics Consortium (IMSGC) for the GWAS data presented in this manuscript (EGAS00000000101), and are grateful to anonymous referees for helpful comments. We also thank Katherine Savage for graphic art. This work was supported by National Institute of Health grant R01NS049477, the National Multiple Sclerosis Society grant RG4680A1 and the Hussman Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Genes and Immunity website .
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Hussman, J., Beecham, A., Schmidt, M. et al. GWAS analysis implicates NF-κB-mediated induction of inflammatory T cells in multiple sclerosis. Genes Immun 17, 305–312 (2016). https://doi.org/10.1038/gene.2016.23
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gene.2016.23
This article is cited by
-
Role of pH-sensing receptors in colitis
Pflügers Archiv - European Journal of Physiology (2024)
-
Novel NF-κB reporter mouse for the non-invasive monitoring of inflammatory diseases
Scientific Reports (2023)
-
The impact of the gut microbiome on extra-intestinal autoimmune diseases
Nature Reviews Immunology (2023)
-
Shared Molecular Signatures Across Zika Virus Infection and Multiple Sclerosis Highlight AP-1 Transcription Factor as a Potential Player in Post-ZIKV MS-Like Phenotypes
Molecular Neurobiology (2023)
-
Prediction of combination therapies based on topological modeling of the immune signaling network in multiple sclerosis
Genome Medicine (2021)
