
Abstract
The intestine is a unique organ inhabited by a tremendous number of microorganisms. Intestinal epithelial cells greatly contribute to the maintenance of the symbiotic relationship between gut microbiota and the host by constructing mucosal barriers, secreting various immunological mediators and delivering bacterial antigens. Mucosal barriers, including physical barriers and chemical barriers, spatially segregate gut microbiota and the host immune system to avoid unnecessary immune responses to gut microbes, leading to the intestinal inflammation. In addition, various immunological mediators, including cytokines and chemokines, secreted from intestinal epithelial cells stimulated by gut microbiota modulate host immune responses, maintaining a well-balanced relationship between gut microbes and the host immune system. Therefore, impairment of the innate immune functions of intestinal epithelial cells is associated with intestinal inflammation.
Similar content being viewed by others
Introduction
The gastrointestinal tract is an organ that takes in food, digests it and absorbs food-derived nutrients. Therefore, exogenous microorganisms, such as bacteria, fungi and viruses, can also enter the gut, accompanying food intake. Some of the microorganisms inhabit the intestine symbiotically and form an ecological community called the gut microbiota. However, intestinal microbiota does not just reside inertly in the gut; rather, it confers vital benefits to the host by digesting dietary fibers to short-chain fatty acids (SCFAs) that can be used as an energy source by the host, synthesizing vitamin B and vitamin K, and metabolizing bile acids. Recent evidence has indicated that intestinal microbiota also influences host immunity by directly interacting with host cells or producing several metabolites, including SCFAs and adenosine triphosphate (ATP).1, 2
Between intestinal environmental factors including gut microbes and host immunity, there exist intestinal epithelial cells and several mucosal barriers covering epithelial cells, such as the mucus layer containing antimicrobial molecules. Intestinal epithelial cells, which include absorptive epithelial cells, goblet cells and Paneth cells, have two major roles, ‘segregation’ and ‘mediation,’ to maintain a healthy relationship between gut microbiota and host immunity.
‘Segregation’ is defined as the separation of the gut microbiota and host immune cells. Intestinal epithelial cells construct two types of mucosal barriers, physical barriers and chemical barriers, to spatially segregate gut microbiota in the intestinal lumen and immune cells in the lamina propria. These barriers prevent conflict between gut microbiota and host immune cells that would result in intestinal inflammation.
‘Mediation’ is defined as the delivery of signals between gut microbes and host immune cells. Intestinal epithelial cells react to gut microbes or their metabolites and produce mediators, including cytokines and chemokines, to induce T-cell immune responses or deliver antigens to antigen-presenting cells (APCs) in lymphoid tissues, contributing to antigen-specific IgA responses and the oral tolerance to food antigens. Activated T cells produce several cytokines, including interleukin (IL)-17 and IL-22, which promote the production of antimicrobial molecules by intestinal epithelial cells to regulate the overgrowth of pathogenic opportunistic microbes.
Inflammatory bowel diseases (IBD) include ulcerative colitis (UC) and Crohn’s disease (CD). IBD involves the chronic inflammation of all or part of the digestive tract. Recent evidence has revealed that the dysfunction of intestinal barriers is one of the causes for IBD development. Indeed, the reduced production of mucus or antimicrobial peptides is observed in some IBD patients, and mice genetically defective in mucosal barrier components show a high sensitivity to intestinal inflammation.
In this review, we focus on the two roles of intestinal epithelial cells, ‘segregation’ and ‘mediation,’ in the maintenance of gut homeostasis and the prevention of intestinal inflammation.
Mucosal barriers constructed by intestinal epithelial cells segregate gut microbes and the host immune system
Intestinal epithelial cells generate various types of barriers to protect the intestinal mucosa from commensal microbes or invading pathogenic microorganisms. These barriers are divided into two subtypes, physical barriers and chemical barriers.
Physical barriers include the mucus layer covering the intestinal mucosa, the glycocalyx on the microvilli of absorptive intestinal epithelial cells, and the cell junctions firmly linking intestinal epithelial cells. These barriers physically inhibit the invasion of the mucosa by intestinal microorganisms.
Mucus is a viscous fluid secreted by goblet cells that is enriched in mucin glycoproteins that form large net-like polymers. Mucus secretion from goblet cells is regulated by the host sensing gut microbes or their metabolites, such as SCFAs or Th2 cytokines, including IL-5 and IL-13.3, 4, 5, 6 In addition, recent studies revealed that the activation of the inflammasome mediated by NOD-like receptor family pyrin domain-containing 6 (NLRP6), a member of the NOD-like receptor family, drives mucus granule exocytosis from goblet cells by promoting autophagy.7
In the large intestine, where there are tremendous numbers of intestinal bacteria, the number of goblet cells is much higher than in the small intestine. Therefore, the mucus layer in the large intestine is very thick. It is composed of two layers, the inner, firm mucus layer and the outer, loose mucus layer.8 These mucus layers are organized by gel-forming MUC2, which is a highly O-glycosylated protein produced by goblet cells. The inner mucus layer is a stratified mucus layer anchored to the intestinal epithelia, which contains polymerized MUC2, and it does not allow microorganisms to easily invade the intestinal epithelia.8 Thus, the inner mucus layer is free from intestinal bacteria. Notably, many bacteria are able to invade the colonic epithelia in Muc2-deficient mice lacking the inner mucus layer.8 The inner mucus layer is converted into the outer mucus layer by the proteolytic processing of polymerized MUC2 by the host or bacteria. The outer mucus layer is inhabited by a large number of intestinal microbes. These microbes utilize polysaccharides of MUC2 as an energy source; therefore, the absence of dietary fiber, which is the main energy source of intestinal bacteria, leads to the expansion of mucin-degrading species, resulting in the increase of inner mucus degradation.9
Regarding the mechanism by which the inner mucus layer segregates intestinal bacteria and epithelial cells in the large intestine, various antimicrobial molecules, including immunoglobulin A (IgA) and the defensin family of proteins, that are transported or produced by intestinal epithelial cells are thought to be involved in the protection against bacterial invasion of the inner mucus layer. However, in the large intestine, there are no Paneth cells, which is a specialized intestinal epithelial cell type that produces antimicrobial molecules; thus, the expression level of antimicrobial molecules is not high compared with that in the small intestine.10 It is unclear what molecules are critical in the segregation between intestinal microbes and intestinal epithelia in the large intestine.
In this context, a recent study identified a highly glycosylated Glycosylphosphatidylinositol (GPI)-anchored protein called Ly6/Plaur domain-containing 8 (Lypd8) as a novel molecule contributing to the segregation of intestinal bacteria and intestinal epithelia in the large intestine.11 Lypd8, which is anchored to the intestinal epithelial cells in the uppermost epithelial layer, is constitutively shed into the intestinal lumen and preferentially binds to flagellated bacteria from genera such as Escherichia, Proteus and Helicobacter, thereby inhibiting the bacterial invasion of the colonic epithelia. Mice lacking Lypd8 demonstrate the disappearance of the bacteria-free space just above the epithelial layer of the colon, indicating that Lypd8 is critical for the segregation of intestinal bacteria and colonic epithelia.11
Even if bacteria can penetrate the inner mucus layer, the glycocalyx, a meshwork of carbohydrate moieties of glycolipids or glycoproteins, including transmembrane mucins, faces invading bacteria as a barrier on the epithelial cell surface. In addition, cell junctions, such as the tight and adhesion junctions linking epithelial cells, physically hamper microbial invasion through the paracellular pathway. Recent reports revealed that the expression of cell junction-associated molecules, including occludins and claudins, is upregulated by indole, a metabolite of dietary tryptophan from commensal bacteria possessing tryptophanase after Pregnane X receptor stimulation.12, 13
In the small intestine, where there are fewer goblet cells than in the large intestine, chemical barriers, including antimicrobial peptides (AMPs) and the regenerating islet-derived 3 (Reg3) family of proteins produced by intestinal epithelial cells, particularly Paneth cells, have critical roles in the segregation of intestinal bacteria and epithelial cells.14, 15 AMPs are small, basic amino acid-rich cationic proteins that are evolutionally conserved in a wide range of organisms. They include the defensin family of proteins and cathelicidins, both of which interact with the negatively charged microbial cell membrane and cause membrane disruption by forming pore-like structures.16 Defensins are classified into α-, β- and θ-defensins, among which α-defensin (cryptdin) mainly protects against pathogenic bacterial infection. Pro-cryptdin is converted into mature-cryptdin by matrix metalloproteinase-7 (MMP-7); therefore, MMP-7-deficient mice lacking mature-cryptdin are highly susceptible to Salmonella typhimurium infection.17
The Reg3 family of proteins was recently identified as novel antimicrobial molecules mainly produced by Paneth cells. In particular, Reg3γ is active against Gram-positive bacteria and has a critical role in the spatial separation of the intestinal bacteria and intestinal epithelia in the small intestine.15, 18, 19
Intestinal epithelial cells express pattern recognition receptors, including Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-containing proteins (NODs). The production of antimicrobial molecules by Paneth cells is regulated by TLR4/MyD88 signaling and NOD2 signaling driven by gut microorganisms.14, 15, 20 In mice lacking NOD2, which recognizes muramyl dipeptides, which are conserved structures in bacterial peptidoglycans, the expression of defensins is substantially reduced, resulting in high susceptibility to Listeria monocytogenes infection.20
Gut immune cells also influence mucosal barriers through the production of cytokines or direct cell–cell contact. IL-17 and IL-22 produced by Th17 cells or type3 innate lymphoid cells (ILC3) upregulate the secretion of AMPs and Reg3 family proteins by intestinal epithelial cells.21 In addition, IL-6 derived from intraepithelial lymphocytes enhances intestinal epithelial cell proliferation and contributes to healing from mucosal injury.22 Conversely, other pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α and interferon (IFN)-γ, inhibit epithelial cell proliferation through the suppression of β-catenin/T cell factor (TCF) signaling.23 In mucosal injury, activated macrophages differentiated from monocytes recruited to the mucosal wound site trigger the colonic epithelial progenitor niche with direct cell–cell contact to promote epithelial regeneration, which helps in the recovery of the mucosal barrier.24 Th2 cytokines, such as IL-5 and IL-13, promote colonic wound healing by inducing the alternative activation of macrophages, which contributes to epithelial cell proliferation.25 In contrast, IL-13, which is upregulated in the colon of patients with UC, has been shown to promote the apoptosis of intestinal epithelial cells, leading to mucosal barrier disturbance.26, 27
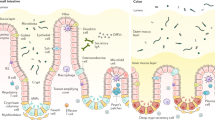
As described above, intestinal epithelial cells produce both physical barriers, such as the mucus layer, glycocalyx and cell junctions and chemical barriers, including AMPs and the Reg3 family of proteins, which are regulated by intestinal environmental factors and immune cell-derived cytokines. These barriers ingeniously segregate commensal microbes and host immune cells to prevent unnecessary conflict between symbiotic microbes and host immune cells, thereby maintaining their symbiotic relationship in the intestine (Figure 1).

Mucosal barriers in the gut. In the small intestine, chemical barriers, including AMPs produced by Paneth cells, have major roles in the segregation of intestinal bacteria and intestinal epithelial cells. In contrast, in the large intestine, which is inhabited by a tremendous number of bacteria, intestinal bacteria and intestinal epithelial cells are separated by the inner mucus layer containing polymerized MUC2. Lypd8, a GPI-anchored protein expressed in the epithelial cells, promotes the segregation of the two by binding to intestinal bacteria, especially flagellated bacteria. AMP, antimicrobial peptides; LZ, lysozyme; TLR, toll-like receptor; ILC3, type 3 innate lymphoid cell.
Intestinal epithelial cells mediate the crosstalk between gut microbes and the host immunity
The intestinal microenvironment, including gut microbiota and their metabolites, is easily and rapidly altered by diet, drugs, stress, infection with bacterial or viral pathogens and even jet lag.28, 29, 30, 31, 32, 33 Thus, host immunity must adapt to alterations in the gut environment, including dysbiosis and pathogenic bacterial infections. Therefore, intestinal epithelial cells stimulated by gut environmental factors interact with host immune cells and modulate gut immune cell responses.
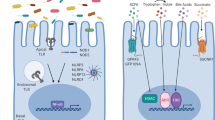
Segmented filamentous bacteria (SFB) are commensal bacteria found in the mouse or rat intestine, most of which attach to intestinal epithelial cells in the ileum. The attachment of SFB strongly facilitates Th17 cell differentiation by inducing the production of serum amyloid A (SAA) by intestinal epithelial cells (Figure 2a).34, 35 In addition, SFB stimulate IL-23 receptor-dependent IL-22 production by activated ILC3.36 Furthermore, the attachment to intestinal epithelial cells of Citrobacter rodentium, an enteric pathogen, upregulates the expression of Nos2, Duoxa2 and Duox2, which are involved in the production of reactive oxygen species, facilitating Th17 cell differentiation in the colonic lamina propria.35 IL-17 and IL-22 from Th17 cells or ILC3 promote the production of antimicrobial molecules, including AMPs and Reg3 family proteins, to control gut microbiota. IL-22 also affects the glycosylation of intestinal epithelial cell-surface proteins by inducing the expression of fucosyltransferase 2 (Fut2).37 The fucosylation of membrane proteins by Fut2 in intestinal epithelial cells is critical for protection against infection by enteric pathogens, such as S. typhimurium.37, 38, 39

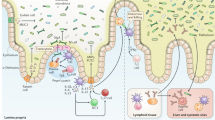
Intestinal epithelial cells mediate the crosstalk between gut microbes and the host immunity. Intestinal epithelial cells stimulated by gut environmental factors modulate gut immune cell responses by secreting humoral factors, such as serum amyloid A (SAA) and several kinds of cytokines (a, b). Intestinal epithelial cells also inactivate bacteria-derived stimulators to host immune cells, such as adenosine triphosphate (ATP), to regulate excessive host immune responses (c). In addition, M cells, which specialize in the uptake and delivery of antigens in the intestinal lumen, contribute to the induction of antigen-specific IgA responses by delivering bacterial or dietary antigens to dendritic cells (DCs) (d).
Intestinal epithelial cells also modulate host immune responses by secreting cytokines and chemokines. The TLR5/MyD88 signaling stimulated by the flagellin proteins of Gram-negative rod bacteria, such as Escherichia and Proteus, promotes the production of IL-8 by epithelial cells, which recruits neutrophils to the lamina propria.40, 41 Recent studies demonstrated that tuft cells, taste-chemosensory epithelial cells, contribute to the elimination of parasites by producing IL-25, which activates ILC2 to secrete IL-13 inducing Th2 responses, resulting in an enhancement of tuft cell and goblet cell differentiation (Figure 2b).42, 43, 44 IL-33, a member of the IL-1 family, which is released from damaged intestinal epithelial cells, promotes Th2 responses by activating ILC2 to produce IL-5 and IL-13 during parasitic infections (Figure 2b).4, 45 In addition, recent studies found that IL-33 from intestinal epithelial cells also promotes the suppressive function of colonic regulatory T (Treg) cells expressing ST2, an IL-33 receptor.46, 47 Thymic stromal lymphopoietin (TSLP), a four-helix bundle cytokine that is produced by intestinal epithelial cells, contributes to protection against helminth infection by inducing Th2 immune responses.48 In addition, intestinal epithelial cells drive IgA class switching in B cells present in the intestinal lamina propria by releasing a proliferation-inducing ligand (APRIL) through TLR-inducible signaling.49
Intestinal epithelial cells also produce mediators other than cytokines and chemokines that modulate gut immune responses. MUC2, which is produced by goblet cells, not only organizes the mucus layer but also constrains the immunogenicity of gut antigens by delivering tolerogenic signals to dendritic cells (DCs).50 Some peptide hormones, including cholecystokinin, glucagon-like peptide (GLP) 1 and 2, and serotonin, are secreted by intestinal endocrine cells in response to luminal nutrients and gut microbiota; they both directly or indirectly affect gut immunity.51 Cholecystokinin regulates the differentiation and cytokine production of CD4+ T cells and B cells, and it also controls macrophage activation by inhibiting inducible nitric oxide synthase (iNOS) production.52, 53, 54, 55 In addition, cholecystokinin indirectly inhibits the release of inflammatory cytokines from macrophages by triggering the secretion of acetylcholine (Ach) from vagal afferents.56
Recent studies revealed that some metabolites produced by commensal microbes directly influence T cell immune responses. ATP derived from commensal bacteria drives Th17 differentiation through the activation of CD70high CD11b+ DCs in the intestine.2 In terms of ATP-dependent Th17 cell differentiation, intestinal epithelial cells regulate excessive Th17 cell activation by controlling the ATP concentration in the intestinal lumen through the expression of ATP-degrading enzymes, such as ecto-nucleoside triphosphate diophosphohydrolase (E-NTPD) 7 (Figure 2c).57
In addition to the production of humoral factors that regulate gut immunity, intestinal epithelial cells can contribute to gut adaptive immune responses by delivering antigens to gut immune cells. M cells, which are found in follicle-associated epithelia, specialize in the uptake and delivery of antigens from the lumen to APCs, including DCs, and have a critical role in the induction of antigen-specific IgA (Figure 2d).58 Glycoprotein 2 (GP2) is a transcytolic receptor on M cells that participates in the uptake of bacterial antigens; therefore, antigen-specific IgA responses are attenuated in mice lacking GP2.59, 60 Recent reports indicate that Spi-B, an Ets transcription factor, is a master regulator for M cell functional and structural maturation, and M cell-dependent T cell activation in Spi-B-deficient mice is impaired.61, 62
Thus, between intestinal environmental factors and gut immunity, intestinal epithelial cells operate as sensors to signals from gut microbes and host immune cells and mediate the balance between both players by secreting cytokines, chemokines and hormones.
Intestinal inflammation caused by the dysfunction of mucosal barriers
IBD is a group of intractable diseases characterized by chronic inflammation of the digestive tract and includes UC and CD. The incidence and prevalence of IBD are increasing with time and in different regions around the world, indicating that the elucidation of the pathogenesis of IBD is a matter of great urgency.63 Recent evidence has revealed that both gut environmental factors and host immune dysfunction based on genetic predisposition contribute to the development of IBD.64 With regard to genetic predisposition underlying the pathogenesis of IBD, recent genome-wide association studies using next-generation sequencing technology identified various IBD susceptibility genes, which include the mucosal barrier-related genes FUT2, MUC19 and NOD2.65, 66, 67, 68 Indeed, the reduced production of mucosal barrier-related molecules, such as AMPs and mucins, is observed in the intestines of IBD patients.69 in addition, the expression of LYPD8, which inhibits bacterial invasion in the colon, was found to be severely decreased in the colonic mucosa of some UC patients.11
Many studies using genetically modified mice with mucosal barrier dysfunction have demonstrated the relationship between mucosal barrier dysfunction and intestinal inflammation. Mice lacking MUC2 are missing the inner mucus layer physically segregating gut microbes and colonic epithelia and develop spontaneous colitis because of the bacterial invasion of the colonic mucosa.8, 70 In accordance with the deficiency of MUC2, the lack of cooperation of core 1 synthase (C1galt), which synthesizes the major constituent of the O-glycan core structure, allows bacteria to penetrate the inner mucus layer, resulting in spontaneous colitis. Mice devoid of Fut2, which mediates the transfer of fucoses to the terminal galactose on glycans in cell-surface glycoproteins, are highly susceptible to S. typhimurium infection.37 In mice deficient in Lypd8, a larger number of flagellated bacteria, such as Proteus and Escherichia, can invade the inner mucus layer, which causes high susceptibility to dextran sulfate sodium (DSS)-induced intestinal inflammation.11 In mice lacking NLRP6, an inflammasome-associated molecule, mucus secretion from goblet cells is impaired, and these mice show the disappearance of the bacteria-free zone in the inner mucus layer and thus are highly sensitive to DSS-induced intestinal inflammation and infection with bacterial pathogens such as Citrobacter rodentium.7, 71 Interestingly, wild-type mice cohoused with NLRP6-deficient mice, which have dysbiosis of the intestine, also show high susceptibility to DSS-induced intestinal inflammation, indicating colitogenic dysbiosis is transmissible between individuals.71 The dysfunction of cell junctions also causes intestinal inflammation. The intestinal deletion of Claudin-7, which is an important component of tight junctions, enhances paracellular organic solute flux and causes colitis in mice.72
The impairment of chemical barriers also causes high susceptibility to intestinal inflammation. Mice lacking MyD88 in intestinal epithelial cells show decreased production of AMPs and mucus by intestinal epithelial cells, and consequently, they are highly susceptible to experimental colitis and enteric bacterial infection.73, 74 Mice deficient in IL-22 that promotes the production of AMPs by intestinal epithelial cells, also show a high sensitivity for DSS colitis.75 In addition, intestinal epithelial cell-specific inhibition of nuclear factor (NF)-κB through the conditional ablation of NEMO, an IκB kinase subunit essential for NF-κB activation, causes the spontaneous development of intestinal inflammation in mice because of the impaired expression of antimicrobial peptides and bacterial translocation into the colonic mucosa.76 Mice deficient in NOD2, which is a susceptibility gene for CD, do not show spontaneous intestinal inflammation but show severe Th1-driven granulomatous inflammation of the ileum induced by Helicobacter hepaticus because of the reduced production of AMPs by Paneth cells.77, 78, 79 Deficiency in adaptor protein (AP)-1B, which mediates the sorting of membrane proteins, causes epithelial immune dysfunction, such as the reduced expression of antimicrobial proteins and the impaired secretion of IgA, leading to chronic colitis with an enhanced Th17 response.80
It has been recently indicated that mucosal barrier dysfunction based on genetic predisposition leads to the alteration of gut microbiota composition, dysbiosis, and enhanced susceptibility to intestinal inflammation and the infection of pathogens. Furthermore, dysbiosis induced by environmental factors, such as a high-fat diet and some medicines, accelerates intestinal inflammation in genetically predisposed individuals (Figure 3).

The imbalance between mucosal barriers and gut microbes promotes susceptibility to intestinal inflammation. Dysfunction of mucosal barriers because of genetic predisposition, such as the decreased production of AMPs and mucin, allows intestinal bacteria to gain access to gut immune cells, thereby contributing to the development of intestinal inflammation. Dysbiosis induced by environmental factors, such as a high-fat diet and various medicines, accelerates intestinal inflammation in situations in which the mucosal barrier is disrupted. AMPs, antimicrobial peptides.
Conclusions
Intestinal epithelial cells create various kinds of mucosal barriers to ‘segregate’ gut microbes and gut immune cells, and sense signals from both populations and secrete humoral factors to ‘mediate’ the balance between both populations, both of which contribute to the maintenance of gut homeostasis. Accordingly, defects in these functions of intestinal epithelial cells in mice promote the development of disorders demonstrating intestinal inflammation, similar to human IBD. Thus, the dysfunction of the intestinal mucosal barrier system is thought to cause IBD. At present, therapies for intractable IBD are limited, and several different therapies, including immunosuppressive treatment, are required. A more detailed knowledge of the mechanisms regulating gut mucosal barrier system will certainly lead to novel therapeutic approaches for IBD.
References
Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013; 504: 446–450.
Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M et al. ATP drives lamina propria T(H)17 cell differentiation. Nature 2008; 455: 808–812.
Kim YS, Ho SB . Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep 2010; 12: 319–330.
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 2010; 463: 540–544.
Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci USA 2010; 107: 11489–11494.
Shimotoyodome A, Meguro S, Hase T, Tokimitsu I, Sakata T . Short chain fatty acids but not lactate or succinate stimulate mucus release in the rat colon. Comp Biochem Physiol A Mol Integr Physiol 2000; 125: 525–531.
Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014; 156: 1045–1059.
Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC . The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA 2008; 105: 15064–15069.
Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 2016; 167: 1339–1353 e21.
Cunliffe RN, Mahida YR . Expression and regulation of antimicrobial peptides in the gastrointestinal tract. J Leukoc Biol 2004; 75: 49–58.
Okumura R, Kurakawa T, Nakano T, Kayama H, Kinoshita M, Motooka D et al. Lypd8 promotes the segregation of flagellated microbiota and colonic epithelia. Nature 2016; 532: 117–121.
Shimada Y, Kinoshita M, Harada K, Mizutani M, Masahata K, Kayama H et al. Commensal bacteria-dependent indole production enhances epithelial barrier function in the colon. PLoS ONE 2013; 8: e80604.
Venkatesh M, Mukherjee S, Wang H, Li H, Sun K, Benechet AP et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014; 41: 296–310.
Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ . Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol 2000; 1: 113–118.
Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011; 334: 255–258.
Brogden KA . Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 2005; 3: 238–250.
Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, Lopez-Boado YS, Stratman JL et al. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science 1999; 286: 113–117.
Cash HL, Whitham CV, Behrendt CL, Hooper LV . Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 2006; 313: 1126–1130.
Mukherjee S, Zheng H, Derebe MG, Callenberg KM, Partch CL, Rollins D et al. Antibacterial membrane attack by a pore-forming intestinal C-type lectin. Nature 2014; 505: 103–107.
Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 2005; 307: 731–734.
Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 2006; 203: 2271–2279.
Kuhn KA, Manieri NA, Liu TC, Stappenbeck TS . IL-6 stimulates intestinal epithelial proliferation and repair after injury. PLoS ONE 2014; 9: e114195.
Capaldo CT, Beeman N, Hilgarth RS, Nava P, Louis NA, Naschberger E et al. IFN-gamma and TNF-alpha-induced GBP-1 inhibits epithelial cell proliferation through suppression of beta-catenin/TCF signaling. Mucosal Immunol 2012; 5: 681–690.
Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS . Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci USA 2005; 102: 99–104.
Seno H, Miyoshi H, Brown SL, Geske MJ, Colonna M, Stappenbeck TS . Efficient colonic mucosal wound repair requires Trem2 signaling. Proc Natl Acad Sci USA 2009; 106: 256–261.
Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005; 129: 550–564.
Heller F, Fromm A, Gitter AH, Mankertz J, Schulzke JD . Epithelial apoptosis is a prominent feature of the epithelial barrier disturbance in intestinal inflammation: effect of pro-inflammatory interleukin-13 on epithelial cell function. Mucosal Immunol 2008; 1 (Suppl 1): S58–S61.
Zhang M, Yang XJ . Effects of a high fat diet on intestinal microbiota and gastrointestinal diseases. World J Gastroenterol 2016; 22: 8905–8909.
Theriot CM, Koenigsknecht MJ, Carlson PE Jr, Hatton GE, Nelson AM, Li B et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 2014; 5: 3114.
Thaiss CA, Zeevi D, Levy M, Zilberman-Schapira G, Suez J, Tengeler AC et al. Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell 2014; 159: 514–529.
Fujisaka S, Ussar S, Clish C, Devkota S, Dreyfuss JM, Sakaguchi M et al. Antibiotic effects on gut microbiota and metabolism are host dependent. J Clin Invest 2016; 126: 4430–4443.
Bailey MT, Dowd SE, Parry NM, Galley JD, Schauer DB, Lyte M . Stressor exposure disrupts commensal microbial populations in the intestines and leads to increased colonization by Citrobacter rodentium. Infect Immun 2010; 78: 1509–1519.
Singh P, Teal TK, Marsh TL, Tiedje JM, Mosci R, Jernigan K et al. Intestinal microbial communities associated with acute enteric infections and disease recovery. Microbiome 2015; 3: 45.
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009; 139: 485–498.
Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 2015; 163: 367–380.
Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell 2015; 163: 381–393.
Goto Y, Obata T, Kunisawa J, Sato S, Ivanov II, Lamichhane A et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science 2014; 345: 1254009.
Pham TA, Clare S, Goulding D, Arasteh JM, Stares MD, Browne HP et al. Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe 2014; 16: 504–516.
Pickard JM, Maurice CF, Kinnebrew MA, Abt MC, Schenten D, Golovkina TV et al. Rapid fucosylation of intestinal epithelium sustains host–commensal symbiosis in sickness. Nature 2014; 514: 638–641.
Yu Y, Zeng H, Lyons S, Carlson A, Merlin D, Neish AS et al. TLR5-mediated activation of p38 MAPK regulates epithelial IL-8 expression via posttranscriptional mechanism. Am J Physiol Gastrointest Liver Physiol 2003; 285: G282–G290.
Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL . Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol 2001; 167: 1882–1885.
Gerbe F, Sidot E, Smyth DJ, Ohmoto M, Matsumoto I, Dardalhon V et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 2016; 529: 226–230.
Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 2016; 351: 1329–1333.
von Moltke J, Ji M, Liang HE, Locksley RM . Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 2016; 529: 221–225.
Humphreys NE, Xu D, Hepworth MR, Liew FY, Grencis RK . IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J Immunol 2008; 180: 2443–2449.
Duan L, Chen J, Zhang H, Yang H, Zhu P, Xiong A et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3(+) regulatory T-cell responses in mice. Mol Med 2012; 18: 753–761.
Schiering C, Krausgruber T, Chomka A, Frohlich A, Adelmann K, Wohlfert EA et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 2014; 513: 564–568.
Taylor BC, Zaph C, Troy AE, Du Y, Guild KJ, Comeau MR et al. TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. J Exp Med 2009; 206: 655–667.
He B, Xu W, Santini PA, Polydorides AD, Chiu A, Estrella J et al. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity 2007; 26: 812–826.
Shan M, Gentile M, Yeiser JR, Walland AC, Bornstein VU, Chen K et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science 2013; 342: 447–453.
Worthington JJ . The intestinal immunoendocrine axis: novel cross-talk between enteroendocrine cells and the immune system during infection and inflammatory disease. Biochem Soc Trans 2015; 43: 727–733.
Li Q, Han D, Cong B, Shan B, Zhang J, Chen H et al. Cholecystokinin octapeptide significantly suppresses collagen-induced arthritis in mice by inhibiting Th17 polarization primed by dendritic cells. Cell Immunol 2011; 272: 53–60.
Saia RS, Mestriner FL, Bertozi G, Cunha FQ, Carnio EC . Cholecystokinin inhibits inducible nitric oxide synthase expression by lipopolysaccharide-stimulated peritoneal macrophages. Mediat Inflamm 2014; 2014: 896029.
Zhang JG, Liu JX, Jia XX, Geng J, Yu F, Cong B . Cholecystokinin octapeptide regulates the differentiation and effector cytokine production of CD4(+) T cells in vitro. Int Immunopharmacol 2014; 20: 307–315.
Zhang JG, Cong B, Li QX, Chen HY, Qin J, Fu LH . Cholecystokinin octapeptide regulates lipopolysaccharide-activated B cells co-stimulatory molecule expression and cytokines production in vitro. Immunopharmacol Immunotoxicol 2011; 33: 157–163.
Luyer MD, Greve JW, Hadfoune M, Jacobs JA, Dejong CH, Buurman WA . Nutritional stimulation of cholecystokinin receptors inhibits inflammation via the vagus nerve. J Exp Med 2005; 202: 1023–1029.
Kusu T, Kayama H, Kinoshita M, Jeon SG, Ueda Y, Goto Y et al. Ecto-nucleoside triphosphate diphosphohydrolase 7 controls Th17 cell responses through regulation of luminal ATP in the small intestine. J Immunol 2013; 190: 774–783.
Mabbott NA, Donaldson DS, Ohno H, Williams IR, Mahajan A . Microfold (M) cells: important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol 2013; 6: 666–677.
Ohno H, Hase K . Glycoprotein 2 (GP2): grabbing the FimH bacteria into M cells for mucosal immunity. Gut Microbes 2010; 1: 407–410.
Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M et al. Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature 2009; 462: 226–230.
Kanaya T, Hase K, Takahashi D, Fukuda S, Hoshino K, Sasaki I et al. The Ets transcription factor Spi-B is essential for the differentiation of intestinal microfold cells. Nat Immunol 2012; 13: 729–736.
Sato S, Kaneto S, Shibata N, Takahashi Y, Okura H, Yuki Y et al. Transcription factor Spi-B-dependent and -independent pathways for the development of Peyer's patch M cells. Mucosal Immunol 2013; 6: 838–846.
Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012; 142: 46–54 e42.
Goto Y, Kurashima Y, Kiyono H . The gut microbiota and inflammatory bowel disease. Curr Opin Rheumatol 2015; 27: 388–396.
Anderson CA, Boucher G, Lees CW, Franke A, D'Amato M, Taylor KD et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet 2011; 43: 246–252.
Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet 2010; 42: 1118–1125.
Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491: 119–124.
Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015; 47: 979–986.
Jager S, Stange EF, Wehkamp J . Inflammatory bowel disease: an impaired barrier disease. Langenbecks Arch Surg 2013; 398: 1–12.
Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006; 131: 117–129.
Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011; 145: 745–757.
Tanaka H, Takechi M, Kiyonari H, Shioi G, Tamura A, Tsukita S . Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut 2015; 64: 1529–1538.
Frantz AL, Rogier EW, Weber CR, Shen L, Cohen DA, Fenton LA et al. Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin-2, and antibacterial peptides. Mucosal Immunol 2012; 5: 501–512.
Bhinder G, Stahl M, Sham HP, Crowley SM, Morampudi V, Dalwadi U et al. Intestinal epithelium-specific MyD88 signaling impacts host susceptibility to infectious colitis by promoting protective goblet cell and antimicrobial responses. Infect Immun 2014; 82: 3753–3763.
Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA . Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 2008; 29: 947–957.
Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007; 446: 557–561.
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 2001; 411: 599–603.
Biswas A, Liu YJ, Hao L, Mizoguchi A, Salzman NH, Bevins CL et al. Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc Natl Acad Sci USA 2010; 107: 14739–14744.
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 2001; 411: 603–606.
Takahashi D, Hase K, Kimura S, Nakatsu F, Ohmae M, Mandai Y et al. The epithelia-specific membrane trafficking factor AP-1B controls gut immune homeostasis in mice. Gastroenterology 2011; 141: 621–632.
Acknowledgements
We thank C Hidaka for secretarial assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Okumura, R., Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp Mol Med 49, e338 (2017). https://doi.org/10.1038/emm.2017.20
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2017.20
This article is cited by
-
Regulation of inflammatory diseases via the control of mRNA decay
Inflammation and Regeneration (2024)
-
METTL3 restricts RIPK1-dependent cell death via the ATF3-cFLIP axis in the intestinal epithelium
Cell Regeneration (2024)
-
Signaling pathways in colorectal cancer: implications for the target therapies
Molecular Biomedicine (2024)
-
Fungal polysaccharides from Inonotus obliquus are agonists for Toll-like receptors and induce macrophage anti-cancer activity
Communications Biology (2024)
-
Death receptor 5 is required for intestinal stem cell activity during intestinal epithelial renewal at homoeostasis
Cell Death & Disease (2024)
