
Abstract
Ethnically specific data on genetic variation are crucial for understanding human biology and for clinical interpretation of variant pathogenicity. We analyzed data obtained by deep sequencing 1303 Korean whole exomes; the data were generated by three independent whole exome sequencing projects (named the KOEX study). The primary focus of this study was to comprehensively analyze the variant statistics, investigate secondary findings that may have clinical actionability, and identify loci that should be cautiously interpreted for pathogenicity. A total of 495 729 unique variants were identified at exonic regions, including 169 380 nonsynonymous variants and 4356 frameshift insertion/deletions. Among these, 76 607 were novel coding variants. On average, each individual had 7136 nonsynonymous single-nucleotide variants and 74 frameshift insertion/deletions. We classified 13 pathogenic and 13 likely pathogenic variants in 56 genes that may have clinical actionability according to the guidelines of the American College of Medical Genetics and Genomics, and the Association for Molecular Pathology. The carrier frequency of these 26 variants was 2.46% (95% confidence interval 1.73–3.46). To identify loci that require cautious interpretation in clinical sequencing, we identified 18 genes that are prone to sequencing errors, and 671 genes that are highly polymorphic and carry excess nonsynonymous variants. The catalog of identified variants, its annotation and frequency information are publicly available (http://koex.snu.ac.kr). These findings should be useful resources for investigating ethnically specific characteristics in human health and disease.
Similar content being viewed by others


Introduction
Technical advances in massive parallel sequencing have resulted in increased application of whole-exome sequencing (WES), not only for research purposes but also for clinical genetic diagnosis. As the United States Food and Drug Administration approved marketing authorization for the first next-generation sequencer in 2013, its clinical application is expected to expand more rapidly.1 It has been reported that WES can provide a potential molecular genetic diagnosis in ~25% of cases referred for suspected genetic disorders in clinical genetics laboratories.2 WES studies are also applied in genomics research on a large scale to identify causal coding variants of complex disorders.3 A comprehensive catalog of ethnically specific genetic variations and its frequency spectrum are crucial for determining variant pathogenicity as well as examining the quality of WES procedures. Currently, there are several genetic variation databases, such as those derived from the 1000 Genomes Project,4 NHLBI Exome Sequencing Project,5 and Exome Aggregation Consortium.6 However, most of these sequencing projects involved European populations, and more genetic information is required for other populations, including East Asians.
When WES is performed, a large number of variants are identified. Variants that are not directly related to the specific condition for which WES is performed are referred to as secondary or incidental findings.7 Some of these exonic variants might be in genes that result in disorders that can be asymptomatic for a long period and can be prevented or treated. The American College of Medical Genetics and Genomics (ACMG) has recommended reporting pathogenic or likely pathogenic variants in a minimum list of 56 genes associated with 24 medical conditions.7 In addition, the ACMG and the Association for Molecular Pathology (AMP) have published standards and guidelines for interpreting the pathogenicity of sequence variation.8 These standards and guidelines consist of 28 attributes for evaluating evidence, including population frequency, functional experiments, familial segregation, computational prediction and rules to classify variants into five categories of pathogenicity. However, Amendola et al.9 have suggested that these criteria need to be clarified to reduce errors in the application of the ACMG-AMP standards and guidelines and discrepancies between laboratories. Although several studies have investigated the incidence of secondary findings in exome sequencing studies,10, 11, 12, 13, 14 secondary findings in East Asian populations have not been thoroughly investigated on a large scale using the strict criteria of the recent ACMG-AMP standards and guidelines.
On the other hand, there are loci that are prone to misinterpretation with regard to variant pathogenicity. These loci could be either (1) susceptible to sequencing errors or (2) highly polymorphic with excess nonsynonymous variants. There have been limited attempts to identify false-positive signals by filtering highly variable genes and to screen loci with excess heterozygosity in WES.15, 16 Comprehensive investigations are required to identify genes that should be cautiously interpreted when WES is performed to identify disease-causing mutations. In addition, it is still unknown how these loci differ depending on the sequencing methods or the ethnicity of the study population. In this KOrean whole EXome sequencing (KOEX) study, we investigated the following: (1) the characteristics of variants identified from 1303 Korean exomes, (2) the secondary findings of 56 ACMG-recommended genes and (3) genes that should be cautiously interpreted for pathogenicity.
Materials and methods
Study design and participants
We investigated exonic variants identified from 1303 participants of a KOEX study. The KOEX study consisted of three individual WES projects. Project 1 was the Seoul National University Hospital type 2 diabetes case–control WES study (SNUH project 1), which consisted of 910 participants. Project 2 consisted of 191 normal healthy parents from the Seoul National University rare disease WES study (SNUH project 2). Project 3 consisted of 202 subjects from the Green Cross hemophilia WES study (Green Cross project). The Institutional Review Board (IRB) of the Seoul National University Hospital (IRB No. H-1205-130-411 for SNUH project 1, H-1406-081-588 for SNUH project 2) and the Green Cross Laboratories (IRB No. GCRL 2014-02) approved the projects, and written informed consent was obtained from each participant. Brief descriptions of each project are shown in Table 1.
WES and variant calling
Table 1 shows the whole-exome capture kit, sequencing system and median coverage information for each project. Paired-end sequence reads were aligned to the human reference genome (GRCh37). All data were processed using BWA,17 Picard software (http://broadinstitute.github.io/picard/), GATK18 pipelines of the Broad Institute to align the sequence reads. Variant calling was performed using GATK HaplotypeCaller in GVCF mode, and CombineGVCF was employed to merge the data into each cohort. GenotypeGVCF and VariantQualityScoreRecalibration (VQSR) were performed as recommended by the developers. ApplyRecalibration was performed with option –ts-filter-level 99.5 for single-nucleotide variants (SNVs) and 99.0 for insertion/deletions (INDELs).
Quality control of WES and variant annotation
To effectively remove sites with low depth (DP), genotype quality (GQ) and call rate, we imposed stringent genotype level filters: variants were called at sites where the DP was ⩾7, the GQ was ⩾20 and the call rate was >0.90. Bi-allelic variants with significant deviation (P<0.001) from the Hardy–Weinberg Equilibrium (HWE) were excluded using VCFtools.19 The remaining variants after filtering were regarded as high-quality variants.
We calculated per-sample metrics, such as total number of variants, number of singleton and doubleton variants, mean X chromosome heterozygosity, mean transition to transversion ratio (Ti/Tv) and ratio of heterozygous to nonreference homozygous sites (Het/Hom), in order to filter-out low-quality samples. Six samples with an excess X chromosome heterozygosity and abnormal Het/Hom ratio were removed. We further estimated inter-individual relatedness by identity-by-state and multi-dimensional scaling analysis using PLINK20 and removed 30 samples. Basic annotation of each variant was undertaken using Annotate Variation (ANNOVAR)21 according to RefSeq gene transcripts. Further detailed annotation was performed using the Human Gene Mutation Database (HGMD) professional version release 2016.122 and the ClinVar database.23
Population structure and principal components analysis
To compare the genetic variation found in our study of Korean participants with that of other populations, we performed population structure and principal component analyses. The Korean data were merged with the 26 populations in release 3 of the 1000 Genomes Project. We combined autosomal SNVs commonly shared in the dataset with minor allele frequency (MAF)>5%. SNVs with genotype missingness>1% were discarded, and linkage disequilibrium-based SNV pruning was performed using the –indep-pairwise option in a sliding window of 50 SNVs moved by 10 SNVs and an r2 threshold of 0.1 in PLINK.24 The final dataset contained 3807 unrelated individuals with 64 626 SNVs. We used a model-based clustering algorithm in ADMIXTURE v1.2325 to examine the genetic structure of the merged dataset. ADMIXTURE identifies K genetic clusters and assigns the proportions of each genotype membership to each cluster. We performed ADMIXTURE analyses considering the K values from 2 to 15, and the model showed best predictive accuracy for K=7. Principal component analysis was performed using GCTA v1.24.726 with the merged data set. We extracted the top eight principal components of the variance-standardized relationship matrix.
Analysis of secondary findings of WES
To investigate the secondary findings of WES, we primarily focused on 56 genes recommended by ACMG.7 Non-silent variants located in these genes were evaluated for pathogenicity according to the standards and guidelines recommended by ACMG-AMP.8 Among all non-silent variants, we selected either low- or high-confidence disease-causing mutations using HGMD.22 We further applied a MAF filter of <0.5% and the evidence for pathogenicity was evaluated for 28 attributes. These data were combined using a scoring rule to classify each variant as either pathogenic (P), likely pathogenic (LP), benign (B), likely benign (LB) or variant of uncertain significance (VUS). Guidelines were strictly followed, and recently suggested modifications were adopted.9 Methods for applying each evidence criterion were similar to a recent report.12 Three investigators reviewed all the evidence attributes for each variant independently and made a consensus agreement for variants with a P or a LP classification. All lines of evidence were manually reviewed for P or LP variants, known pathogenic variants, and truncating variants. Detailed methods for variant pathogenicity classification are described in Supplementary Methods. We calculated the allele count and carrier frequency (95% confidence interval (CI)) of P or LP variants in our study participants. The CI of carrier frequency was calculated using a modified Wald method.27
Identification of loci prone for misinterpretation
We evaluated loci that are prone to misinterpretation and compiled a list of genes that should be provisionally excluded when investigating disease-causing mutations. First, we listed genes with low-quality protein coding DNA sequence (CDS) variants that were filtered during quality control procedures such as GATK-VQSR and HWE tests. We defined genes with (1) more than 100 variants filtered by VQSR or (2) with more than five coding variants with significant deviation from HWE (P<0.001), as genes susceptible for sequencing errors. Second, we identified highly polymorphic loci by evaluating the per-gene metric for the nonsynonymous variant burden for each sample, considering the allele frequency and gene size in combination. Using the longest transcript of each gene, 10 714 RefSeq genes on autosomes with CDS lengths longer than 1 kb were evaluated. Genes containing more nonsynonymous variants than the third (upper) quartile+1.5 × (interquartile range) in terms of numbers or rates (number of variants divided by the length of the transcript) were considered to have excess nonsynonymous variants, according to the outlier detection method of Tukey.28 We then further defined highly polymorphic genes if the gene showed excess nonsynonymous variants in at least two of the following four categories: (1) excess absolute number of entire nonsynonymous variants, (2) excess absolute number of rare (MAF<0.5%) nonsynonymous variants, (3) excess rate of entire nonsynonymous variants and (4) excess rate of rare nonsynonymous variants. The same rule was also applied to the 1000 Genomes Projects to validate the misinterpretable genes suggested in this study.
Results
Characteristics of variants identified by WES
A total of 1303 participants from three WES projects were included in the KOEX study. Brief descriptions of the three WES projects are shown in Table 1. After filtering low-quality samples and variants, we identified 495 729 unique variants at exonic regions (Table 2). The number of variants that were identified in each WES project and their overlap are displayed in Supplementary Figure 1. A total of 293 048 variants were located at CDS (169 380 nonsynonymous, 1665 splicing, 3642 stop gain/loss, 107 148 synonymous SNVs, and 4356 frameshift and 3221 in-frame INDELs). On average, each individual had 7136 nonsynonymous SNVs and 74 frameshift INDELs located in CDS. In addition, there was on average 177 variants that were predicted to result in protein truncation (splicing, stop gain/loss and frameshift). Further detailed information regarding the frequency distribution of INDELs with regard to its length, singleton and doubleton variant counts according to exonic variant annotation is shown in Supplementary Figure 1. There was a relatively large overlap between variants identified in this study and those of East Asian participants of the Exome Aggregation Consortium (59.1% of SNVs) and East Asian participants of the 1000 Genomes Project (34.7% of SNVs). We identified 76 607 novel coding variants (73 241 SNVs and 3366 INDELs) not cataloged in dbSNP build 147. Most of these variants were very rare (singleton: 87.0%, doubleton: 9.2%) or rare (MAF<0.5%: 99.8%). Population stratification and principal component analyses showed that Korean participants clustered with East Asians of the 1000 Genomes Project and were separated from other populations (Figure 1).29, 30

Population stratification and principle component analyses. The merged data of 1303 Korean participants and the 1000 Genomes Project were investigated (a) for population structure analysis using the ADMIXTURE and (b) for principal component analysis. The Korean participants of our study clustered with East Asians and were separated from other populations. ACB, African Caribbeans in Barbados; AFR, African; AMR, American; ASW, Americans of African Ancestry in SW USA; BEB, Bengali from Bangladesh; CDX, Chinese Dai in Xishuangbanna, China; CEU, Utah Residents (CEPH) with Northern and Western Ancestry, USA; CHB, Han Chinese in Beijing, China; CHS, Southern Han Chinese; CLM, Colombians from Medellin, Colombia; EAS, East Asian; ESN, Esan in Nigeria; EUR, European; FIN, Finnish in Finland; GBR, British in England and Scotland, UK; GIH, Gujarati Indian from Houston, Texas, USA; GWD, Gambian in Western Divisions in the Gambia; IBS, Iberian Population in Spain; ITU, Indian Telugu from the UK; JPT, Japanese in Tokyo, Japan; KHV, Kinh in Ho Chi Minh City, Vietnam; KOR, Korean; LWK, Luhya in Webuye, Kenya; MSL, Mende in Sierra Leone; MXL, Mexican Ancestry from Los Angeles, USA; PEL, Peruvians from Lima, Peru; PJL, Punjabi from Lahore, Pakistan; PUR, Puerto Ricans from Puerto Rico; SAS, South Asian; STU, Sri Lankan Tamil from the UK; TSI, Toscani in Italy; YRI, Yoruba in Ibadan, Nigeria.
Secondary findings in the 56 ACMG recommended genes
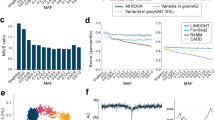
A total of 1049 unique non-silent variants (1004 SNVs and 45 INDELs) were identified in 53 out of the 56 ACMG recommended genes (Supplementary Table 1). On average, there were 19.8 non-silent variants per gene for genes with at least one non-silent variant. For each individual, there was on average 28.7 non-silent variants in these 53 genes. Among 1049 variants, 150 SNVs had a population MAF less than 0.5% and were reported as disease-causing mutations (with either high or low confidence) in HGMD (Supplementary Table 2). An additional 34 INDELs had an MAF <0.5%. These 184 variants were further analyzed for pathogenicity using the recent ACMG-AMP guidelines for variant interpretation. After stringent application of the guidelines and manual review of the evidence, we classified 13 variants as P with an allele count of 15 and a carrier frequency of 1.15% (95% CI 0.68–1.91%) as shown in Table 3. In addition, 13 variants were classified as LP with an allele count of 17 and a carrier frequency of 1.30% (95% CI 0.80–2.10%). Altogether, the carrier frequency of P or LP variants was 2.46% (95% CI 1.73–3.46). Among the 26 P or LP variants, 18 (70.4%) were non-silent SNVs and 8 (29.6%) were frameshift INDELs. All the P or LP variants were singletons, except for variants in MLH1, MYL3, RYR2 and SCN5A, which were observed twice or thrice in our study participants. P variants were identified in BRCA1, BRCA2, DSC2, MLH1, MSH2, MYBPC3, MYL3 and SCN5A. LP variants were identified in KCNH2, LDLR, MYBPC3, MYL3, RYR2 and SCN5A. Each of the 28 evidence attributes that were invoked for the 26 P or LP variants are displayed in Figure 2. The P or LP variants were referenced in ClinVar, and the concordance for actionability (P or LP) was 36.6%.

Evidence attributes for the 26 P or LP variants in 56 ACMG genes. Individual evidence attributes of the 26 variants classified as P or LP are shown. Exonic functions of the variants are shown on the right. Classification was based on the ACMG guidelines (Calls) or determined using the ClinVar database. ACMG, American College of Medical Genetics and Genomics.
Loci prone to misinterpretation of pathogenicity
Next, with the advantage of our sample size, we investigated loci whose pathogenicity is prone to misinterpretation and, therefore, require further scrutiny when evaluating pathogenicity (Figure 3). First, loci that are susceptible to sequencing errors due to low genotype quality or excess heterozygosity were evaluated by performing VQSR and HWE tests. We identified 11 genes with excess variants filtered by VQSR, which include MUC6, MUC3A, ZNF717, MUC16, AHNAK2, FLG, HYDIN, PLIN4, MUC17, PDE4DIP and CDC27 (Supplementary Table 3). Most of these genes were present in segmental duplications and had excess coverage and allelic imbalance. There were also 12 genes with multiple variants deviated from HWE (P<0.001), including ZNF717, PDE4DIP, HYDIN, TPTE, MUC6, OR4K1, PRAMEF2, SCGB1C1, MUC17, HNRNPCL2, PRIM2 and OR8U1 (Supplementary Table 4). These genes were mostly located at/near centromeric or telomeric regions. Second, highly polymorphic genes were evaluated by assessing the burden of nonsynonymous variants. The excess of nonsynonymous variants was estimated using per-gene, per-sample statistics using a combination of burden (number versus rate) and allele frequency (entire versus rare variant). Genes with excess numbers of nonsynonymous variants that were relatively large were associated with extracellular matrix organization and the cytoskeleton (data not shown). By contrast, genes with excess rates of nonsynonymous variants that were mostly small were associated with olfactory transduction, keratin filament and the plasma membrane. We externally validated these findings using the 1000 Genomes Project data (Supplementary Table 5) and observed that most of these genes also showed a similar increased burden in at least one population from the 1000 Genomes Project (Figure 3c–f and Supplementary Figure 2). Of those polymorphic genes in each category, 671 genes that were found in at least two of the four categories were listed as highly polymorphic genes, as shown in Supplementary Table 5.

Characterization of highly misinterpretable loci. (a) Example of a sequencing error prone gene, MUC6, showing excess coverage (gray) and imbalanced allelic fraction (blue) of VQSR filtered variants. (b) Distribution of variants with significant deviation from HWE (green), indicating sequencing error prone loci. (c–f) The frequency distribution of genes according to the burden of nonsynonymous variants and their cutoff values for highly polymorphic genes. The cutoff values for (c) excess number of entire nonsynonymous variants was 1.88, (d) excess rate of entire nonsynonymous variants was 0.78 × 10−3, (e) excess number of rare nonsynonymous variants was 0.064 and (f) excess rate of rare nonsynonymous variants was 0.027 × 10−3. The dashed line indicates the cut-off value for each category. The Venn diagrams show how the highly polymorphic genes in Koreans overlap with the 1000 Genomes Project (1KGP). VQSR, Variant Quality Score Recalibration.
Discussion
In this KOEX study, we comprehensively investigated genetic variations in a Korean population by analyzing the data obtained using high-quality whole-exome deep sequencing. Among the 1303 participants, we identified 495 729 unique exonic variants, of which 73 241 and 3366 were novel coding SNVs and INDELs, respectively. There were on average 7136 nonsynonymous SNVs and 74 frameshift INDELs per individual. Among the 1049 non-silent variants in the 56 ACMG recommended genes, we reviewed 184 rare variants that were suggested to be associated with Mendelian disorders. We identified 13 P and 13 LP variants within these genes with a carrier frequency of 2.46%. Most of these variants were singletons, and 29.6% were INDELs. Finally, we created a list of genes that require cautious interpretation due to a high rate of sequencing error or being highly polymorphic. The variant information, including annotation and allele frequency, is publically available (http://koex.snu.ac.kr). To the best of our knowledge, this is one of the largest studies to investigate genetic variants at the whole-exome scale in Korean populations. The information provided herein should be a useful resource for understanding human biology and discovering novel disease mutations.
One of the primary objectives of this study was to generate a comprehensive high-quality catalog of exonic variations in Koreans. In this study, SNUH project 1, which comprised the majority of the samples, was sequenced with a median coverage of 103.7X, and the remaining two projects were also sequenced with a median coverage of more than 50 ×. We also used a stringent quality control filter to generate high-quality variant calls. Ethnically specific genetic variation data are important, especially for rare protein coding variants. It is well known that exonic variants are rare, and rare variants are mostly confined to closely related populations.4 Among the 293 048 CDS variants identified in the KOEX study, 26.1% were not cataloged in dbSNP build 147 and were regarded as novel. Furthermore, we found that more than 90% of protein truncating variants were novel and were absent in other population databases. A recent study showed that WES in isolated populations could be useful not only for discovering rare Mendelian disease genes but also for identifying rare deleterious variants of common complex disorders.31 We hope that this ethnically specific rare variant information will spur on the identification of causative genetic variation of both rare and common disorders in Koreans.
When WES is performed for clinical or research purposes, reporting secondary findings with clinical actionability is recommended. Among the 56 genes recommended by the ACMG, we categorized 26 variants as either P or LP. The carrier frequency of these P or LP variants was 2.46%. This finding is comparable to other studies that reported a 2–4% carrier frequency in other populations.10, 11, 12 One recent study involving 196 Korean exomes reported a 6.6% carrier frequency of P or LP variants.13 However, the study was limited in sample size and might have over-estimated the frequency. The estimated carrier frequency of P or LP variants in clinically actionable genes could differ depending on several factors, such as the list of genes used, the population on which the study was performed, and specific methods by which the guidelines were applied. We confined our analyses to the 56 ACMG recommended genes, and strictly applied the ACMG-AMP standards and guidelines for variant classification. To obtain data comparable to other studies, when we applied the guidelines we also tried to follow the specific details as published in recent studies.9, 12 Whether carrier frequency varies depending on ethnicity and, if so, by how much, is unknown. There are suggestions that certain populations might have been underrepresented in the literature and in clinical genetics databases.10 As we filtered SNVs that were annotated in HGMD as disease-causing mutations for further clinical interpretation of pathogenicity, we might have underestimated the carrier frequency in our study population. Furthermore, a significant proportion of our participants were patients at SNUH, which is one of the largest tertiary hospitals in Korea. These participants might have complex medical presentations, and it is possible that our estimation could be different from that of the general population.
There could be several reasons why false positive genotypes are obtained when WES is performed.15 Even after these sequencing errors are excluded, there could still be loci that are highly polymorphic and have an excess of nonsynonymous variants. Here, we presented genes that should be cautiously interpreted as disease causing. Loci that were prone to sequencing error were evaluated by genotype quality (VQSR) and excess of heterozygosity (HWE). We further listed genes that were highly polymorphic by evaluating the number or rate of nonsynonymous variants (either entire or rare variants) per-gene for each sample. We provide a full list of genes that were filtered using these methods. The specific cut-off values used to define sequencing error prone genes and highly polymorphic genes were arbitrary. However, genes listed as prone to sequencing error had characteristics of segmental duplication, excess coverage, and excess allelic imbalance. Furthermore, genes listed as being highly polymorphic in our study were associated with categories that include olfactory receptors and keratin filaments known to contain multiple nonsynonymous variants.15 To evaluate whether the list of highly polymorphic genes was valid, we applied the same filter to the 1000 Genomes Project data and found a similar increased burden of nonsynonymous variants in the identified genes. It would be useful to further narrow down specific regions within the listed genes for cautious interpretation of pathogenicity.
There are certain limitations to this study. First, the sample size was modest. The lowest detectable MAF in our study was 0.038%. Nevertheless, this is one of the first major studies to investigate high-quality deep sequenced variants in East Asians. In addition, this is the largest WES study of Korean populations. Second, different whole-exome capture kits were used for each of the three projects. Although the target region covered for each project was different, most of the coding region in which we were primarily interested overlapped for the three capture kits. Third, large INDELs that might be important for certain Mendelian disorders were not included in this study. Methods for identifying large INDELs in WES are still incomplete. Further improvements and validations are required for these methods.
In conclusion, we have evaluated genetic variants in 1303 Korean exomes and provide ethnically specific variation information, including novel rare functional variants. The proportion of P or LP variants in our study population was estimated to be 2.46%, which was comparable to the levels reported for other populations. Finally, we suggest a method for filtering genes that are prone to sequencing errors or that are highly polymorphic with excess nonsynonymous variants. We also provide a list of genes that require caution when interpreting disease causality. As WES is being increasingly used for both clinical and research purposes, our KOEX study results should serve as a valuable resource, especially regarding the exclusion of false positive findings and identifying true disease-causing pathogenic variants. Furthermore, large-scale sequencing studies are expected to broaden the catalog of rare variants and should accelerate the realization of precision medicine.
References
Collins FS, Hamburg MA . First FDA authorization for next-generation sequencer. N Engl J Med 2013; 369: 2369–2371.
Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014; 312: 1870–1879.
Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V, Gaulton KJ et al. The genetic architecture of type 2 diabetes. Nature 2016; 536: 41–47.
The 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM et al. A global reference for human genetic variation. Nature 2015; 526: 68–74.
Fu W, O’Connor TD, Jun G, Kang HM, Abecasis G, Leal SM et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 2013; 493: 216–220.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536: 285–291.
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013; 15: 565–574.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–423.
Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD et al. Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet 2016; 98: 1067–1076.
Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, Gallego CJ et al. Actionable, pathogenic incidental findings in 1000 participants’ exomes. Am J Hum Genet 2013; 93: 631–640.
Amendola LM, Dorschner MO, Robertson PD, Salama JS, Hart R, Shirts BH et al. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res 2015; 25: 305–315.
Maxwell KN, Hart SN, Vijai J, Schrader KA, Slavin TP, Thomas T et al. Evaluation of ACMG-Guideline-Based Variant Classification of Cancer Susceptibility and Non-Cancer-Associated Genes in Families Affected by Breast Cancer. Am J Hum Genet 2016; 98: 801–817.
Jang M-A, Lee S-H, Kim N, Ki C-S . Frequency and spectrum of actionable pathogenic secondary findings in 196 Korean exomes. Genet Med 2015; 17: 1–5.
Gambin T, Jhangiani SN, Below JE, Campbell IM, Wiszniewski W, Muzny DM et al. Secondary findings and carrier test frequencies in a large multiethnic sample. Genome Med 2015; 7: 54.
Fuentes Fajardo KV, Adams D, Program NCS, Mason CE, Sincan M, Tifft C et al. Detecting false-positive signals in exome sequencing. Hum Mutat 2012; 33: 609–613.
Adams DR, Sincan M, Fuentes Fajardo K, Mullikin JC, Pierson TM, Toro C et al. Analysis of DNA sequence variants detected by high-throughput sequencing. Hum Mutat 2012; 33: 599–608.
Li H, Durbin R . Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al. The variant call format and VCFtools. Bioinformatics 2011; 27: 2156–2158.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Wang K, Li M, Hakonarson H . ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164.
Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN . The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 2014; 133: 1–9.
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014; 42: D980–D985.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ . Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015; 4: 7.
Alexander DH, Novembre J, Lange K . Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009; 19: 1655–1664.
Yang J, Lee SH, Goddard ME, Visscher PM . GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011; 88: 76–82.
Agresti A, Coull BA . Approximate is better than ‘exact’ for interval estimation of binomial proportions. Am Stat 1998; 52: 119–126.
Tukey JW Exploratory Data Analysis: Past, Present and Future. Defense Technical Information Center; Fort Belvoir, VA, USA, 1993.
Wong LP, Lai JK, Saw WY, Ong RT, Cheng AY, Pillai NE et al. Insights into the genetic structure and diversity of 38 South Asian Indians from deep whole-genome sequencing. PLoS Genet 2014; 10: e1004377.
Wong LP, Ong RT, Poh WT, Liu X, Chen P, Li R et al. Deep whole-genome sequencing of 100 southeast Asian Malays. Am J Hum Genet 2013; 92: 52–66.
Lim ET, Wurtz P, Havulinna AS, Palta P, Tukiainen T, Rehnstrom K et al. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet 2014; 10: e1004494.
Acknowledgements
This research was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI15C1595, HI14C0060, HI15C3131, HI13C2148 and HI13C1468).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Kwak, S., Chae, J., Choi, S. et al. Findings of a 1303 Korean whole-exome sequencing study. Exp Mol Med 49, e356 (2017). https://doi.org/10.1038/emm.2017.142
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2017.142
This article is cited by
-
Central neurocytoma exhibits radial glial cell signatures with FGFR3 hypomethylation and overexpression
Experimental & Molecular Medicine (2024)
-
Genomic analysis as a tool to infer disparate phylogenetic origins of dysembryoplastic neuroepithelial tumors and their satellite lesions
Scientific Reports (2023)
-
An integrative approach for exploring the nature of fibroepithelial neoplasms
British Journal of Cancer (2023)
-
Frequency of actionable secondary findings in 7472 Korean genomes derived from the National Project of Bio Big Data pilot study
Human Genetics (2023)
-
A database of 5305 healthy Korean individuals reveals genetic and clinical implications for an East Asian population
Experimental & Molecular Medicine (2022)
