
Abstract
Increased oxidative stress has an important role in asthmatic airway inflammation and remodeling. A potent methyl donor, S-adenosylmethionine (SAMe), is known to protect against tissue injury and fibrosis through modulation of oxidative stress. The aim of this study was to evaluate the effect of SAMe on airway inflammation and remodeling in a murine model of chronic asthma. A mouse model was generated by repeated intranasal challenge with ovalbumin and Aspergillus fungal protease twice a week for 8 weeks. SAMe was orally administered every 24 h for 8 weeks. We performed bronchoalveolar lavage (BAL) fluid analysis and histopathological examination. The levels of various cytokines and 4-hydroxy-2-nonenal (HNE) were measured in the lung tissue. Cultured macrophages and fibroblasts were employed to evaluate the underlying anti-inflammatory and antifibrotic mechanisms of SAMe. The magnitude of airway inflammation and fibrosis, as well as the total BAL cell counts, were significantly suppressed in the SAMe-treated groups. A reduction in T helper type 2 pro-inflammatory cytokines and HNE levels was observed in mouse lung tissue after SAMe administration. Macrophages cultured with SAMe also showed reduced cellular oxidative stress and pro-inflammatory cytokine production. Moreover, SAMe treatment attenuated transforming growth factor-β (TGF-β)-induced fibronectin expression in cultured fibroblasts. SAMe had a suppressive effect on airway inflammation and fibrosis in a mouse model of chronic asthma, at least partially through the attenuation of oxidative stress and TGF-β-induced fibronectin expression. The results of this study suggest a potential role for SAMe as a novel therapeutic agent in chronic asthma.
Similar content being viewed by others


Introduction
Asthma, which is defined as chronic airway inflammation and airway hyper-responsiveness, affects approximately 300 million people worldwide.1, 2 Most patients show a favorable response to inhaled corticosteroids (ICS); however, progressive respiratory symptoms and a decline in lung function are observed in some asthmatics. Airway remodeling, which consists of mucus gland hypertrophy, increased airway smooth muscle mass and subepithelial fibrosis, is one of the defining features of chronic asthma. To date, no asthma drug has showed satisfactory effects on airway remodeling.3 Therefore, the development of novel drugs that target both chronic inflammation and fibrosis is an important unmet need in asthma management.
Oxidative stress due to the imbalance between oxidative forces and the antioxidant defense system is considered a critical factor in the induction of chronic asthma. Excess reactive oxygen species (ROS) are reported to enhance inflammatory cell recruitment, pro-inflammatory cytokine production and the accumulation of extracellular matrix proteins in the airway wall in several animal studies.4, 5 Additionally, enhanced oxidative stress is significantly associated with decreased lung function, which is one of the important clinical features of severe asthma.6, 7, 8 The precise mechanism underlying the induction of irreversible airway obstruction by excessive ROS has not been clarified. However, oxidative stress is known to augment airway remodeling by stimulating the production of transforming growth factor-β1 (TGF-β1), fibronectin and vascular endothelial growth factor during lung fibroblasts.9, 10, 11
S-adenosylmethionine (SAMe) is a principal biological methyl donor and key mediator of glutathione and polyamine synthesis that has a crucial role in many biochemical processes.12 The therapeutic effects of SAMe in acute liver injury and liver fibrosis were well demonstrated through many experimental models and clinical trials.13, 14, 15, 16 These effects are known be related to reduced oxidative stress, the maintenance of mitochondrial function, regulation of the cell cycle and inhibition of various mediators that have important roles in the development of liver inflammation and fibrosis.17, 18 Recently, several studies with SAMe have been conducted in various diseases, including lung fibrosis.19, 20, 21, 22, 23 Therefore, it was conceivable that SAMe might have a role in the suppression of airway inflammation and reduction in airway remodeling. However, a therapeutic role for SAMe in asthma had not been investigated previously. Using an inhalational model of Aspergillus fumigatus-induced chronic asthma, we demonstrated anti-inflammatory and antifibrotic effects of SAMe on allergic airway inflammation, most likely through the reduction of oxidative stress and inhibition of TGF-β1 signaling pathway activation.
Materials and methods
Animals and treatments
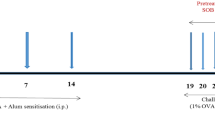
Six-to-eight-week-old female BALB/c mice were maintained in a pathogen-free area. To generate the chronic asthma model, the mice were intranasally challenged twice a week for 8 weeks with 22 μg of chicken ovalbumin (OVA; Grade V; Sigma Chemical Co., St Louis, MO, USA) and 5 μg fungal protease (protease from Aspergillus melleus; Sigma). In the control group, phosphate-buffered saline was administered. The animals were divided into three groups as follows: sham, a low dose of SAMe (30 mg kg−1; Sigma), and a high dose of SAMe (150 mg kg−1). SAMe was administered daily with feedings for 8 weeks starting at day 0 (Figure 1). All mice were killed at day 58, and lung tissue was obtained for histopathological examination. The animal experiments described in this study were approved by our Institutional Animal Care and No Endobronchial Lesion Use Committee.

Experimental protocol for generating the murine model of chronic asthma. Female BALB/c mice were intranasally challenged with 22 μg ovalbumin (OVA) and 8 μg fungal-associated allergenic protease (FAP) twice a week for 8 weeks. S-adenosylmethionine (SAMe) was administered daily via the intraoral route according to the following dosages: 0, 30 and 150 mg kg−1 for 8 weeks. The mice were killced 2 days after the last challenge.
Bronchoalveolar lavage (BAL) fluid analysis
Mice were anesthetized and a trachea was cannulated to prepare for the procedure. BAL was performed with 2 ml phosphate-buffered saline through a tracheal cannula. BAL fluid samples were centrifuged, and the cell pellets were resuspended. Total cell counts were enumerated and differential cell counts were determined in a cytospin sample stained with Diff-Quick (Sysmex. Hyogo, Japan).
Histopathological evaluation
For histopathological examination, lungs were fixed in 10% neutral-buffered formalin for 24 h, embedded in paraffin wax, cut into 4-μm-thick sections and stained with hematoxylin and eosin (H&E). The degree of lung tissue inflammation was scored on a scale of 0–4 (0: none, 1: minimal, 2: mild, 3: moderate, and 4: marked). Additionally, periodic acid-Schiff (PAS) was used to identify mucus production, and Masson’s trichrome staining was used to detect lung fibrosis.
Cell culture
Peribronchial and axillary lymph nodes (LNs) were used to determine various cytokine levels in the animal model. These were obtained aseptically and trimmed of excess fat. The nodes were homogenized with a 25-gauge syringe tip to obtain a single-cell suspension that was then washed twice in phosphate-buffered saline and centrifuged at 1500 g for 10 min at 4 °C. Cell pellets were resuspended in RPMI 1640 (Welgene, Daegu, Korea) with 10% fetal bovine serum and 1% penicillin/streptomycin (Welgene). Cell suspensions were dispended into a 48-well plate, with each well receiving 4 × 105 cells in 0.5 ml of media. The cells were stimulated with 500 μg ml−1 of OVA per well, and the supernatants were harvested 72 h after stimulation. To evaluate the underlying mechanisms of SAMe function, human macrophage (U937) and fibroblast (MRC5) cell lines were used. U937 cells were cultured in RPMI 1640 with 10% fetal bovine serum and 1% penicillin/streptomycin to 90% confluency. Cell suspensions were dispended into 100-mm culture dishes (Corning, Tokyo, Japan) at a density of 5 × 106 cells/dish and pretreated with 10 ng ml−1 of phorbol myristate acetate (Invivogen, San Diego, CA, USA) for 24 h. After 24 h, 75 μM SAMe was added, and cells were maintained for another 24 h. The cells were then resuspended in a 48-well plate, with each well receiving 1 ml of medium containing 1 × 105 cells. The cells were stimulated with either 10 μg ml−1 lipopolysaccharide (LPS; Sigma) or 10 μg ml−1 DerP extract (Yonsei University, Seoul, Korea) for 24 h after SAMe treatment.
MRC5 cells were cultured in Minimum Essential Medium (Welgene) with 10% fetal bovine serum (Gibco, Grand Island, NY, USA), 1% penicillin/streptomycin and 1 mM sodium pyruvate (Welgene) to 90% confluency. The MRC-5 cells were seeded at a density of 2 × 105 cells per plate in a 60-mm dish and incubated overnight. After a 10-μM SAMe pretreatment, the cells were stimulated with 5 ng TGF-β1 (R&D Systems, Minneapolis, MN, USA) for 24 h.
Enzyme-linked immunosorbent assay
Cytokine concentrations were measured from mice lung-draining LN culture supernatants (described in the ‘In vitro culture’ section) and U937 cell culture supernatants. All cytokines in this study, including interleukin (IL)-4, IL-5, IL-6, IL-10, IL-13, IL-17, interferon (IFN)-γ and TGF-β1, were measured with Duoset ELISA Development Kits (R&D Systems) according to the manufacturer’s instructions.
ROS detection by fluorescence plate reader
U937 cells were dispended into a 96-well plate at a density of 2 × 105 per well in 0.1 ml media and pretreated with or without SAMe for 24 h. Next, the cells were treated with 100 μM hydrogen peroxide and stained with 5 μM 2′,7′-dichlorofluorescein (Abcam, Cambridge, UK) for 30 min. The stained cells were assessed in a fluorescent plate reader (Perking Elmer-Wallac 1420 Victor 2 Multilabel plate reader, PerkinElmer, MA, USA).
Immunoblot analysis
Mouse lung tissue and MRC5 cells from each experiment were lysed in lysis buffer containing protease inhibitors on ice for 20 min. The lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (GE Healthcare, Amersham, UK). The following primary antibodies were used: anti-4-hydroxyl-2-nonenal (anti-HNE; Alpha Diagnostic, San Antonio, TX, USA), anti-β-actin (Bioworld, Dublin, OH, USA), antifibronectin (Abcam), anti-SMAD3, anti-phospho-Smad3 (S423/S425), anti-JNK (anti-c-Jun N-terminal kinase), anti-phospho-JNK (Thr183/Tyr185), anti-ERK (anti-extracellular signal–regulated kinase), and anti-phospho-ERK (Tyr204). All secondary antibodies were purchased from Bethyl Laboratories (Montgomery, TX, USA). The protein bands were detected with an enhanced chemiluminescence ()solution (GenDEPOT, Barker, TX, USA).
Statistical analysis
The results were expressed as the means±s.e.m. and analyzed with the one-way analysis of variance component of the Kruskal–Wallis H-test according to their distribution normality using the SPSS software program version 18.0 (SPSS Inc., Chicago, IL, USA). A P-value<0.05 was considered to be statistically significant.
Results
SAMe attenuates allergic airway inflammation, airway fibrosis, lung oxidative stress and the production of various cytokines in a murine model of chronic asthma
First, to verify whether the chronic inflammatory features were well reflected in our mouse model of chronic asthma in this study, we compared the histopathological findings from the current study to those from our previous studies that used a mouse model of conventional acute asthma. Briefly, the conventional acute asthma model was generated via intraperitoneal injections of 20 μg of chicken OVA with 100 μl alum on days 0 and 14 for sensitization, followed by inhalation challenges with 5% OVA on days 21, 22 and 23 for 30 min.24 The mouse model of chronic asthma that was utilized in this study, which was challenged with a fungal protease and OVA for a period of 8 weeks, produced markedly increased mucus in the airway epithelium, as determined by PAS staining. Additionally, subepithelial collagen deposition was significantly increased in the chronic asthma mouse model compared with that in conventional acute asthma mouse models that were utilized in previous studies (Figures 2a and b).

Histological features of chronic asthma in mouse lung tissue from fungal protease-challenged mice. (a) Periodic acid-Schiff (PAS) staining (lung tissue, original magnification, × 400). (b) Masson’s trichrome staining (lung tissue, original magnification, × 200).
Next we compared the histological feature changes after SAMe administration in the mouse model of chronic asthma. In the H&E-stained lung sections in the chronic asthma model, peribronchial and perivascular cell infiltration and alveolar and vascular congestion were significantly increased compared with the control mice. After SAMe treatment, the magnitude of these parameters decreased remarkably compared with the untreated asthmatic group. SAMe suppressed airway inflammation in a dose-dependent manner (Figures 3a and b). The results of the BAL fluid analysis revealed that the SAMe treatment led to a significant decrease in the number of total BAL cells and in each of the cell types evaluated with the differential cell count, including eosinophils (Figures 3c and d). However, the PAS stain showed no significant difference in mucus cell hyperplasia and the areas of acidic mucus production between the SAMe-treated and untreated asthmatic mice (Figures 4a and b). Subepithelial collagen deposition was measured, and the SAMe-treated lung tissue was found to have a markedly ameliorated degree of collagen deposition in a dose-dependent manner (Figures 4c and d). Additionally, the SAMe-treated asthmatic mice showed lower levels of HNE, a representative marker of lipid peroxidation, in their lung tissue (Figures 5a and b).

Effects of SAMe on airway inflammation. (a) Hematoxylin and eosin staining of lung tissue (original magnification, × 200). (b) Inflammatory scores in the lung tissue. (c) Total cell numbers in the BAL fluid. (d) Differential cell counts in the BAL fluid. The data represent the mean±s.e.m.; control group, n=4; chronic asthma group, n=10. CA, chronic asthma. *P<0.05 compared with the untreated CA group.

Effects of SAMe on airway remodeling. (a) Periodic acid-Schiff (PAS) staining (lung tissue, original magnification, × 400). (b) The degree of mucin production, which was deduced from PAS staining. (c) Masson’s trichrome staining (lung tissue, original magnification, × 200). (d) The degree of fibrosis, as measured by Masson’s trichrome staining, was analyzed by quantitative morphometry using the imaging analysis software; control group, n=4; chronic asthma group, n=10. CA, chronic asthma. *P<0.05 compared with the untreated CA group.

Effects of SAMe on oxidative stress regulation. (a) Western blotting analysis of 4-hydroxyl-2-nonenal (HNE) in lung tissues. (b) Degree of HNE production from western blotting analysis; control group, n=4; chronic asthma group, n=10. HNE. *P<0.05 compared with the untreated CA group.
Several cytokines were measured from lung-draining LNs and lung tissues. In the lung-draining LNs, the IL-5, IL-13 and IL-10 levels were increased in asthmatic mice, whereas IFN-γ levels were not detected. After SAMe administration, all of the cytokine levels were markedly reduced (Figure 6). The results of the cytokine levels measured in the lung tissues that were obtained from each study group showed a similar tendency to those in the LN cultures. The IFN-γ levels in the lung tissue were also decreased after SAMe treatment, although there was no statistical significance (Supplementary Figure S1). However, in the BAL fluid analysis, cytokine levels were below the detectable range (data not shown).

The effects of SAMe on cytokine production in lung-drainage lymph nodes of chronic asthmatic mice (quantified by enzyme-linked immunosorbent assay). (a) IL-5. (b) IL-13. (c) IL-10. The data represent the mean±s.e.m.; control group, n=4; chronic asthma group, n=10. CA, chronic asthma. *P<0.05 compared with the untreated CA group.
SAMe reduces the production of various inflammatory mediators and oxidative stress in cultured human macrophages
A marked increase in the production of cytokines, such as IL-6 and TGF-β1, was observed in LPS or house dust mite (HDM) extract-stimulated U937 cells compared with the control group. After SAMe treatment for 24 h, the cytokine levels were significantly reduced (Figures 7a and b). Next the magnitude of ROS production after hydrogen peroxide stimulation was serially measured. The results showed significantly decreased ROS levels in the SAMe-treated U937 cells at each time point. These antioxidative effects were most prominent within 15 min of stimulation (Figure 7c).

Effects of SAMe on cytokine and reactive oxygen species (ROS) production in U937 cells. (a, b) Levels of cytokine production in human macrophages, as determined with enzyme-linked immunosorbent assays. (a) Human IL-6 (hIL-6). (b) Human TGF-β1 (h TGF-β1). The data represent the mean±s.e.m. PMA, phorbol myristate acetate; LPS, lipopolysaccharide; HDM, house dust mite. *P<0.05 compared with the untreated group. (c) The degree of ROS production in U937 cells. The cells were pretreated with SAMe for 24 h and treated with 100 μM of hydrogen peroxide for 0, 5, 15, 30, 45 and 60 min. ROS were measured using a fluorescent plate reader.
SAMe decreases TGF-β1-induced fibronectin expression by blocking Smad3, ERK and JNK phosphorylation in human fibroblasts
We next confirmed that SAMe significantly reduced TGF-β1-induced fibronectin expression in the human fibroblast cell line, MRC-5 (Figures 8a and b). To elucidate the underlying signaling pathway from the SAMe effects, we evaluated the expression of TGF-β1-induced Smad3 phosphorylated (p-Smad3) for the canonical pathway and phosphorylated JNK (p-JNK) and ERK (p-ERK) for the non-canonical pathway. In the MRC-5 cell culture experiments, the p-Smad3, p-JNK and p-ERK levels were highly expressed within 5 min of TGF-β1 stimulation, and they were diminished by 15 min. The p-Smad3 levels were maintained for more than 30 min. The SAMe pretreated MRC-5 cells showed reduced TGF-β1-induced p-Smad3, p-JNK and p-ERK expression levels (Figures 8c and d).

SAMe reduces airway fibrosis by inhibiting TGF-β1-induced SMAD, JNK and ERK phosphorylation. (a, b) Fibronectin expression in MRC-5 cells pretreated with 10 μM SAMe for 24 h, with 5 ng ml−1 of TGF-β1 added before the analysis. (c, d) Western blots of p-SMAD3, SMAD3, p-JNK, JNK, p-ERK and ERK in MRC-5 cells that were pretreated with 10 μM SAMe for 24 h and treated with 5 ng ml−1 TGF-β1 for 0, 5, 15 and 30 min. All results were expressed as ratios with α-tubulin or β-actin (loading control).
Discussion
In the current study, SAMe effectively suppressed both airway inflammation and airway remodeling in a murine model of chronic asthma. The results revealed that SAMe reduced airway inflammation, excessive oxidative-stress burden and airway fibrosis through inhibiting both Smad-dependent and independent TGF-β1 signaling pathways. These findings suggest that SAMe is potentially an attractive, novel therapeutic agent for controlling chronic asthma.
SAMe, a well-known biological methyl donor generated in the cytosol of every mammalian cell, is linked with several essential metabolic pathways. As a potent antioxidant, SAMe has shown to ameliorate tissue inflammation and fibrosis in multiple disease models.19, 20, 21, 25 As studies on the pathogenesis of severe asthma improved our understanding of the disease, oxidative stress is now considered to be an important factor in the development of airway inflammation and remodeling.9, 10, 26 Based on these facts, we evaluated the role of SAMe in an asthma disease model for the first time.
Consistent with previous research, the current study demonstrated that SAMe treatment effectively reduced tissue infiltration of inflammatory cells in the airway in a dose-dependent manner.17, 20, 22 Furthermore, we observed a decrease in eosinophilic inflammation, which is a hallmark of T helper type 2 (Th2) inflammation. Previous studies demonstrated that SAMe inhibited Th1-dominant inflammation through the suppression of cytokines, such as IL-6, IL-8, tumor necrosis factor-α, and IFN-γ.20, 22, 25 In this study, we identified a significant decrease in not only IFN-γ but also Th2 cytokines, such as IL-5 and IL-13 after SAMe treatment. Taken together, the results of this study support the hypothesis that SAMe generally attenuates airway inflammation via suppression of the Th2 immune response as well as Th1 inflammation.
Interestingly, the levels of IL-10, which is regarded as an anti-inflammatory cytokine, were increased in chronic asthma model and then reduced after SAMe administration. It was reported that IL-10 levels are decreased in asthma and that this phenomenon is associated with a reduction in IL-10-producing regulatory T cells. Additionally, our finding is opposite of what was observed in LPS-induced RAW mouse macrophages.27, 28, 29 However, a reduction in IL-10 levels after SAMe treatment was also shown in a recent report by Li et al.,22 in which SAMe inhibited IL-10 expression in colon cancer cells. TGF-β, which is a pleiotropic cytokine with immunomodulatory property, was reported to induce IL-10 secretion from macrophages or regulatory T cells in several studies.30, 31, 32 Therefore, we speculate that the decreased IL-10 levels in our chronic asthma model was associated with decreased TGF-β, as we observed a reduction of TGF-β in LPS/HDM-stimulated macrophages after SAMe treatment. Further studies are needed to clarify the exact mechanism.
We also observed that SAMe significantly inhibited the levels of IL-6 and TGF-β in LPS/HDM-stimulated macrophages. Previous studies demonstrated that decreased IL-6, tumor necrosis factor-α and TGF-β levels were associated with an anti-inflammatory, antifibrotic response following SAMe administration.22, 23, 25, 33 Although asthma is typically a Th2 immunity-dominant disease, the underlying mechanism of asthma is much more complex than the simple Th2 paradigm. Cytokines, such as IL-6 and TGF-β, are involved in naive T-cell differentiation into Th17 cells, which is thought to induce neutrophil airway inflammation, a hallmark of severe asthma.34 Moreover, TGF-β is one of the main mediators involved in airway remodeling in the asthmatic lung.26
In the current study, SAMe produced an inhibitory effect on subepithelial collagen deposition, one of the key features of airway remodeling, which consists of airway fibrosis and mucus hypersecretion. Airway inflammation itself is a well-known causative factor of airway fibrosis, and our study focused on this aspect; however, it should also be kept in mind that airway fibrosis is not a simple consequence of inflammation, but a distinctive process that occurs in the asthmatic airway.35, 36, 37 TGF-β is a profibrotic cytokine and has an essential role in airway fibrosis. TGF-β is known to signal primarily via intracellular Smad proteins, but several non-Smad signaling cascades exist, including the mitogen-activated protein kinase–ERK, JNK and p38 pathways.38, 39 We found that SAMe reduced TGF-β1-induced phosphorylation of Smad3, JNK and ERK in cultured fibroblasts. Our results suggest that SAMe may reduce airway fibrosis by inhibiting both Smad-dependent and -independent TGF-β1 signal pathways. However, a recent study has reported inconsistent results that SAMe exhibited antifibrotic effects by suppressing TGF-β1-stimulated Sp1-ERK1/2 coupled elements, and not by the Smad-associated pathway, in a liver fibrosis model.33 The exact mechanism underlying the SAMe-induced antifibrotic effect is still unclear; further research is definitely needed.
To the contrary, SAMe showed no effect on mucus hypersecretion in this study, despite the decreased levels of IL-13, which is well known to induce mucus production.40 Previously, Chu et al.41 reported enhanced expression of TGF-β2, but not of TGF-β1, in asthmatic bronchial epithelial cells, which suggests that TGF-β2 is more likely to be involved with mucus hyperproduction. To clarify the underlying mechanism of the differential effect of SAMe on each airway remodeling component, further studies should be carried out. Consistent with results from previous studies,17, 21, 23, 25 our study also showed an antioxidative capacity for SAMe in lung tissue of asthmatic mice and in vitro experiments using human macrophages. Oxidative stress is known to have a critical role in the initiation and aggravation of inflammation as well as airway remodeling.42 Interestingly, it was reported that oxidative stress aggravates Th2-skewed inflammation.43 These results suggest that the reduction of oxidative stress might be more effectively suppressed during Th2-dominant inflammation than other types of inflammation. It is conceivable that the anti-inflammatory and antifibrotic effects of SAMe in this study may be attributed to the antioxidant capacity of SAMe by modulating the overall inflammatory response, especially Th2-dominant inflammation. Nevertheless, we did not separately assess the impact of oxidative stress on Th2 inflammation. To clarify this issue, further studies are warranted.
In the current study, we assessed the therapeutic role of SAMe in chronic airway inflammation and remodeling. A. fumigatus is a potent indoor fungal allergen and a number of previous studies demonstrated that intranasal inhalation of aerosolized A. fumigatus allergen significantly enhances persistent airway hyper-reactivity, mucus cell hyperplasia and peribronchial collagen deposition, which are major features of chronic allergic airway disease.44, 45 Thus we developed a chronic asthma model that included fungal allergen inhalation, which mimicked chronic inflammation and fibrosis much better than our conventional acute asthma model, and confirmed the therapeutic effect of SAMe.
In summary, to the best of our knowledge, our present study is the first to demonstrate a suppressive effect of SAMe on airway inflammation and remodeling in an experimental model of chronic asthma. The observed anti-inflammatory and antifibrotic effects might have been mediated by reduced oxidative stress. Our findings suggest a potential benefit of SAMe as a novel therapeutic drug that targets both airway inflammation and remodeling in chronic asthma. Importantly, SAMe has the advantage of currently being in use with no safety concerns.
References
Bateman ED, Hurd SS, Barnes PJ, Bousquet J, Drazen JM, FitzGerald M et al. Global strategy for asthma management and prevention: GINA executive summary. Eur Respir J 2008; 31: 143–178.
National Asthma Education and Prevention Program. Expert Panel Report 3 (EPR-3): Guidelines for the Diagnosis and Management of Asthma-Summary Report 2007. J Allergy Clin Immunol 2007; 120 (Suppl): S94–S138.
Berair R, Brightling CE . Asthma therapy and its effect on airway remodelling. Drugs 2014; 74: 1345–1369.
Baek KJ, Cho JY, Rosenthal P, Alexander LEC, Nizet V, Broide DH . Hypoxia potentiates allergen induction of HIF-1α, chemokines, airway inflammation, TGF-β1, and airway remodeling in a mouse model. Clin Immunol 2013; 147: 27–37.
Fujieda S, Diaz-Sanchez D, Saxon A . Combined nasal challenge with diesel exhaust particles and allergen induces in vivo IgE isotype switching. Am J Respir Cell Mol Biol 1998; 19: 507–512.
Katsoulis K, Kontakiotis T, Leonardopoulos I, Kotsovili A, Legakis IN, Patakas D . Serum total antioxidant status in severe exacerbation of asthma: correlation with the severity of the disease. J Asthma 2003; 40: 847–854.
Ercan H, Birben E, Dizdar EA, Keskin O, Karaaslan C, Soyer OU et al. Oxidative stress and genetic and epidemiologic determinants of oxidant injury in childhood asthma. J Allergy Clin Immunol 2006; 118: 1097–1104.
Yoon SY, Kim TB, Baek S, Kim S, Kwon HS, Lee YS et al. The impact of total antioxidant capacity on pulmonary function in asthma patients. Int J Tuberc Lung Dis 2012; 16: 1544–1550.
Bellocq A, Azoulay E, Marullo S, Flahault A, Fouqueray B, Philippe C et al. Reactive oxygen and nitrogen intermediates increase transforming growth factor-beta1 release from human epithelial alveolar cells through two different mechanisms. Am J Respir Cell Mol Biol 1999; 21: 128–136.
Qi S, den Hartog GJ, Bast A . Superoxide radicals increase transforming growth factor-beta1 and collagen release from human lung fibroblasts via cellular influx through chloride channels. Toxicol Appl Pharmacol 2009; 237: 111–118.
Sugiura H, Ichinose M, Tomaki M, Ogawa H, Koarai A, Kitamuro T et al. Quantitative assessment of protein-bound tyrosine nitration in airway secretions from patients with inflammatory airway disease. Free Radic Res 2004; 38: 49–57.
Martinez-Lopez N, Varela-Rey M, Ariz U, Embade N, Vazquez-Chantada M, Fernandez-Ramos D et al. S-adenosylmethionine and proliferation: new pathways, new targets. Biochem Soc Trans 2008; 36: 848–852.
Cave M, Deaciuc I, Mendez C, Song Z, Joshi-Barve S, Barve S et al. Nonalcoholic fatty liver disease: predisposing factors and the role of nutrition. J Nutr Biochem 2007; 18: 184–195.
Ko K, Yang H, Noureddin M, Iglesia-Ara A, Xia M, Wagner C et al. Changes in S-adenosylmethionine and GSH homeostasis during endotoxemia in mice. Lab Invest 2008; 88: 1121–1129.
Mato JM, Alvarez L, Ortiz P, Pajares MA . S-adenosylmethionine synthesis: molecular mechanisms and clinical implications. Pharmacol Ther 1997; 73: 265–280.
Yang H, Ko K, Xia M, Li TW, Oh P, Li J et al. Induction of avian musculoaponeurotic fibrosarcoma proteins by toxic bile acid inhibits expression of glutathione synthetic enzymes and contributes to cholestatic liver injury in mice. Hepatology 2010; 51: 1291–1301.
Mato JM, Lu SC . Role of S-adenosyl-L-methionine in liver health and injury. Hepatology 2007; 45: 1306–1312.
Purohit V, Abdelmalek MF, Barve S, Benevenga NJ, Halsted CH, Kaplowitz N et al. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: summary of a symposium. Am J Clin Nutr 2007; 86: 14–24.
Choi LJ, Huang JS . A pilot study of S-adenosylmethionine in treatment of functional abdominal pain in children. Altern Ther Health Med 2013; 19: 61–64.
Feld JJ, Modi AA, El-Diwany R, Rotman Y, Thomas E, Ahlenstiel G et al. S-adenosyl methionine improves early viral responses and interferon-stimulated gene induction in hepatitis C nonresponders. Gastroenterology 2011; 140: 830–839.
Fuso A, Nicolia V, Ricceri L, Cavallaro RA, Isopi E, Mangia F et al. S-adenosylmethionine reduces the progress of the Alzheimer-like features induced by B-vitamin deficiency in mice. Neurobiol Aging 2012; 33: 1482.e1–16.
Li TW, Yang H, Peng H, Xia M, Mato JM, Lu SC . Effects of S-adenosylmethionine and methylthioadenosine on inflammation-induced colon cancer in mice. Carcinogenesis 2012; 33: 427–435.
Sueblinvong V, Kerchberger VE, Saghafi R, Mills ST, Fan X, Guidot DM . Chronic alcohol ingestion primes the lung for bleomycin-induced fibrosis in mice. Alcohol Clin Exp Res 2014; 38: 336–343.
Lee T, Kwon HS, Bang BR, Lee YS, Park MY, Moon KA et al. Grape seed proanthocyanidin extract attenuates allergic inflammation in murine models of asthma. J Clin Immunol 2012; 32: 1292–1304.
Au AY, Hasenwinkel JM, Frondoza CG . Hepatoprotective effects of S-adenosylmethionine and silybin on canine hepatocytes in vitro. J Anim Physiol Anim Nutr (Berl) 2013; 97: 331–341.
Halwani R, Al-Muhsen S, Al-Jahdali H, Hamid Q . Role of transforming growth factor-beta in airway remodeling in asthma. Am J Respir Cell Mol Biol 2011; 44: 127–133.
Eusebio M, Kraszula L, Kupczyk M, Kuna P, Pietruczuk M . The effects of interleukin-10 or TGF-beta on anti-CD3/CD28 induced activation of CD8+CD28- and CD8+CD28+ T cells in allergic asthma. J Biol Regul Homeost Agents 2013; 27: 681–692.
Kawayama T, Matsunaga K, Kaku Y, Yamaguchi K, Kinoshita T, O'Byrne PM et al. Decreased CTLA4(+) and Foxp3(+) CD25(high)CD4(+) cells in induced sputum from patients with mild atopic asthma. Allergol Int 2013; 62: 203–213.
Song Z, Uriarte S, Sahoo R, Chen T, Barve S, Hill D et al. S-adenosylmethionine (SAMe) modulates interleukin-10 and interleukin-6, but not TNF, production via the adenosine (A2) receptor. Biochim Biophys Acta 2005; 1743: 205–213.
Joetham A, Takeda K, Taube C, Miyahara N, Matsubara S, Koya T et al. Naturally occurring lung CD4(+)CD25(+) T cell regulation of airway allergic responses depends on IL-10 induction of TGF-beta. J Immunol 2007; 178: 1433–1442.
Presser K, Schwinge D, Wegmann M, Huber S, Schmitt S, Quaas A et al. Coexpression of TGF-beta1 and IL-10 enables regulatory T cells to completely suppress airway hyperreactivity. J Immunol 2008; 181: 7751–7758.
Maeda H, Kuwahara H, Ichimura Y, Ohtsuki M, Kurakata S, Shiraishi A . TGF-beta enhances macrophage ability to produce IL-10 in normal and tumor-bearing mice. J Immunol 1995; 155: 4926–4932.
Nieto N, Cederbaum AI . S-adenosylmethionine blocks collagen I production by preventing transforming growth factor-beta induction of the COL1A2 promoter. J Biol Chem 2005; 280: 30963–30974.
Nembrini C, Marsland BJ, Kopf M . IL-17-producing T cells in lung immunity and inflammation. J Allergy Clin Immunol 2009; 123: 986–994 quiz 95–96.
Lowe LA, Simpson A, Woodcock A, Morris J, Murray CS, Custovic A . Wheeze phenotypes and lung function in preschool children. Am J Respir Crit Care Med 2005; 171: 231–237.
Saglani S, Payne DN, Zhu J, Wang Z, Nicholson AG, Bush A et al. Early detection of airway wall remodeling and eosinophilic inflammation in preschool wheezers. Am J Respir Crit Care Med 2007; 176: 858–864.
Kariyawasam HH, Aizen M, Barkans J, Robinson DS, Kay AB . Remodeling and airway hyperresponsiveness but not cellular inflammation persist after allergen challenge in asthma. Am J Respir Crit Care Med 2007; 175: 896–904.
Massague J, Blain SW, Lo RS . TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000; 103: 295–309.
Imamichi Y, Waidmann O, Hein R, Eleftheriou P, Giehl K, Menke A . TGF beta-induced focal complex formation in epithelial cells is mediated by activated ERK and JNK MAP kinases and is independent of Smad4. Biol Chem 2005; 386: 225–236.
Zhen G, Park SW, Nguyenvu LT, Rodriguez MW, Barbeau R, Paquet AC et al. IL-13 and epidermal growth factor receptor have critical but distinct roles in epithelial cell mucin production. Am J Respir Cell Mol Biol 2007; 36: 244–253.
Chu HW, Balzar S, Seedorf GJ, Westcott JY, Trudeau JB, Silkoff P et al. Transforming growth factor-β2 induces bronchial epithelial mucin expression in asthma. Am J Pathol 2004; 165: 1097–1106.
Moreno-Macias H, Romieu I . Effects of antioxidant supplements and nutrients on patients with asthma and allergies. J Allergy Clin Immunol 2014; 133: 1237–1244 quiz 45.
King MR, Ismail AS, Davis LS, Karp DR . Oxidative stress promotes polarization of human T cell differentiation toward a T helper 2 phenotype. J Immunol 2006; 176: 2765–2772.
Hogaboam CM, Blease K, Mehrad B, Steinhauser ML, Standiford TJ, Kunkel SL et al. Chronic airway hyperreactivity, goblet cell hyperplasia, and peribronchial fibrosis during allergic airway disease induced by Aspergillus fumigatus. Am J Pathol 2000; 156: 723–732.
Schuh JM, Hoselton SA . An inhalation model of allergic fungal asthma: Aspergillus fumigatus-induced inflammation and remodeling in allergic airway disease. Methods Mol Biol 2013; 1032: 173–184.
Acknowledgements
This study was supported by a grant to YSC from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI14C2628 and HI13C196).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Yoon, SY., Hong, G., Kwon, HS. et al. S-adenosylmethionine reduces airway inflammation and fibrosis in a murine model of chronic severe asthma via suppression of oxidative stress. Exp Mol Med 48, e236 (2016). https://doi.org/10.1038/emm.2016.35
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2016.35
This article is cited by
-
SAM protects against alveolar septal cell apoptosis in autoimmune emphysema rats
European Journal of Medical Research (2023)
-
Asthmatic condition induced the activity of exosome secretory pathway in rat pulmonary tissues
Journal of Inflammation (2021)
-
Characterization of macrophage phenotype, redox, and purinergic response upon chronic treatment with methionine and methionine sulfoxide in mice
Amino Acids (2020)
-
Betaine effects against asthma-induced oxidative stress in the liver and kidney of mice
Molecular Biology Reports (2020)
-
Metabolomic changes in the mouse retina after optic nerve injury
Scientific Reports (2018)

{kind=link}