
Abstract
The epithelial–mesenchymal transition (EMT) is important for embryonic development and the formation of various tissues or organs. However, EMT dysfunction in normal cells leads to diseases, such as cancer or fibrosis. During the EMT, epithelial cells are converted into more invasive and active mesenchymal cells. E-box-binding proteins, including Snail, ZEB and helix–loop–helix family members, serve as EMT-activating transcription factors. These transcription factors repress the expression of epithelial markers, for example, E-cadherin, rearrange the cytoskeleton and promote the expression of mesenchymal markers, such as vimentin, fibronectin and other EMT-activating transcription factors. Signaling pathways that induce EMT, including transforming growth factor-β, Wnt/glycogen synthase kinase-3β, Notch and receptor tyrosine kinase signaling pathways, interact with each other for the regulation of this process. Although the mechanism(s) underlying EMT in cancer or embryonic development have been identified, the mechanism(s) in embryonic stem cells (ESCs) remain unclear. In this review, we describe the underlying mechanisms of important EMT factors, indicating a precise role for EMT in ESCs, and characterize the relationship between EMT and ESCs.
Similar content being viewed by others


Introduction
The epithelial–mesenchymal transition (EMT) is important for generating multiple tissues during organismal development. This process is particularly essential for the gastrulation of metazoans and neural crest delamination of vertebrates.1 EMT is also involved in wound healing. However, EMT dysfunction leads to pathological conditions, including carcinogenesis and fibrosis. The most critical difference between embryonic and tumorigenic EMT is that genetically abnormal cells are employed during EMT for tumorigenesis, and these cells lose sensitivity to normal growth regulatory signals.2 During EMT, polarized epithelial cells are converted into mesenchymal cells. Therefore, epithelial cells lose characteristics that enable differentiation, including cell–cell adhesion, apical–basal polarity and motility dysfunction, and obtain mesenchymal properties, such as motility, invasiveness and apoptotic resistance.3 The results of studies conducted since the 2007 EMT conference held in Poland have suggested several characteristics for the classification of EMT; consequently, three types of EMT were proposed at Cold Spring Harbor Laboratories in 2008.3 Type 1 EMT affects embryo formation, implantation, organ development and the generation of various cell types. Type 2 EMT promotes wound healing, tissue regeneration and fibrosis, and participates in inflammation.3, 4 Type 3 EMT is involved in cancer progression and metastasis.3, 4
Embryonic stem cells (ESCs) are acquired from the inner cell mass of early blastocysts. Accordingly, these cells differentiate into multiple cell types, and can be used to treat diseases through the generation of tissues and organs.5 Mouse and human ESCs have been used for EMT-related research,6 and many differences in the cellular properties of these cells have also been identified (Table 1). For example, human ESCs grow more slowly and have a flattened morphology, different gene expression profile and distinct pluripotent and differentiation signals compared with murine ESCs.7, 8, 9 Therefore, terms such as ‘primed’ and ‘naïve’ have been used to classify these cell types. Naïve cells can be more undifferentiated than primed cells, as naïve cells can be harvested from the inner cell mass of early blastocysts in mouse ESCs, whereas primed cells have been identified in the epiblast of late blastocysts in mouse ESCs. Naïve cells can be subcultured as single cells, forming round colonies. In addition, naïve stem cells possess two active X chromosomes and express stage-specific embryonic antigen-1 on the cell surface.10 The mechanism(s) underlying EMT during cancer or embryonic development have been elucidated. However, the corresponding mechanism(s) of ESCs remains unclear. In this review, we highlight the functions of important EMT factors to determine the precise role for EMT in ESCs and characterize the relationship between EMT and ESCs.
Key EMT signaling molecules
E-cadherin
Cadherins are calcium ion-dependent glycoproteins expressed on the cell surface. These proteins are involved in cell–cell adhesion and interaction. The cadherin family is divided into type 1 and type 2.11 E-cadherin is a cadherin family member that possesses a single-pass transmembrane domain, and this protein is primarily detected in epithelial cells. Although the region of E-cadherin that participates in cell–cell adhesion is unknown, the histidine-alanine-valine domain might have an important role in cell–cell interaction.11, 12 E-cadherin has two preserved domains: the β-catenin-binding domain and p120-binding domain. The β-catenin-binding domain promotes the interaction between the actin cytoskeleton and E-cadherin. This interaction is achieved through the cytoplasmic cell adhesion complex, which comprises epithelial protein lost in neoplasm, β-catenin and α-catenin.11, 13 As decreased E-cadherin expression on the cell surface might be involved in tumor progression and metastasis, E-cadherin is considered a repressor of tumor progression and metastasis.11 Decreased E-cadherin expression breaks down cell–cell contact and increases EMT induction, resulting in tumor motility. For example, in colorectal carcinoma, which expresses STAT3, the expression of E-cadherin was decreased. However, the induction of small interfering RNA for STAT3 increased E-cadherin expression, thereby decreasing the invasive properties and expression of vimentin and N-cadherin in these cells.14 E-cadherin has also been associated with transcriptional repressive pathways through the E-box-binding proteins, Snail and Slug, and matrix metalloproteinase-7 (MMP-7) and MMP-13. For example, MMP-7 and MMP-13 can remove the extracellular E-cadherin domain, and the deleted soluble ectodomain might suppress E-cadherin activity in neighboring cells.11, 15, 16
Transcription factors that regulate EMT
A number of transcription factors, including Snail, ZEB and helix–loop–helix (HLH) family members, regulate EMT.17, 18, 19 In vertebrates, the Snail family comprises three members: Snail 1, Snail 2 and Snail 3.20 Snail 1 is also called ‘Snail,’ and Snail 2 is also known as ‘Slug,’ and these proteins suppress the expression of epithelial genes, such as E-cadherin and plakoglobin, and also activate the expression of mesenchymal proteins, including N-cadherin and fibronectin. The activation of the Snail family depends on a conserved zinc finger domain and an N-terminal snail/Gfi domain.16, 20, 21 In addition, the C-terminal region attaches to the E-box, represses the expression of target genes associated with epithelial cell markers and activates mesenchymal protein expression.20, 21 Other transcription factors, including Ets-1 and specificity protein 1,22, 23 are also involved in this process. The Snail family members are important factors for EMT regulation. In colon cancer (DLD1), cells transfected with Snail and Slug show increased β-catenin –T-cell factor-4 transcription complex formation.24 This complex increases the expression of transforming growth factor (TGF)-β3, resulting in the TGF-β3-induced upregulation of lymphoid enhancer factor-1 gene expression, which induces the EMT response. In addition, TGF-β1 and 2 stimulate EMT signaling pathways through Snail and Slug.24 Snail blocks epithelial markers, that is, cytokeratin-18, Muc-1, desmoplakin and E-cadherin, thereby increasing the expression of fibronectin and vimentin. It upregulates Rho-GTPase during gastrulation, resulting in cytoskeletal changes.16 In lung cancer, Slug acts as a metastasis-promoting gene, demonstrating that slug downregulates E-cadherin expression, upregulates MMP-2 and increases angiogenesis.16
HLH family members also influence the EMT process.20 This protein family can be divided into seven groups. Class 1 HLH proteins include E12 and E47, class 2 HLH proteins include twist and class 5 HLH proteins include Ids. These three HLH groups, classes 1, 2, 5, are involved in EMT induction. E12 or E47 inhibit the expression of E-cadherin and plakoglobin and increase the production of vimentin and fibronectin.20 Ids repress transcription through different molecules, such as TGF-β, associated with the repression of E-cadherin expression.20 Ids do not bind DNA, but rather bind to E12 or E47, acting as negative inhibitors.20, 25 The ectopic expression of twist represses the expression of E-cadherin, occludin and claudin-7 and facilitates the expression of vimentin and N-cadherin to enhance cancer cell migration and invasion.26 Twist forms a heterodimer with E12 and E47, and this dimer regulates transcription through DNA binding.27
In vertebrates, the ZEB family includes ZEB1 (deltaEF1 or AREB6) and ZEB2 (smad-interacting protein 1, SIP1).18 ZEB family proteins possess a zinc finger cluster at each end, which interacts with DNA. A repressor motif in the central homeodomain and the recruitment of C-terminal-binding protein as a co-repressor mediate transcriptional repression through ZEB1 and ZEB2.20 However, the interaction between ZEB, PCAF and p300 converts ZEB1 from a repressor to an activator. These proteins reduce epithelial marker expression and increase mesenchymal marker expression during the induction of EMT.25 ZEB is activated through other signaling molecules, such as TGF-β, and growth factors that activate Ras/mitogen-activated protein kinase; in addition, this protein is repressed through microRNA 200 and microRNA 250 family members.17, 28, 29
Signaling pathways that influence EMT
Several pathways regulate the EMT, including pathways involving TGF-β, Wnt, receptor tyrosine kinase (RTK), hedgehog, tumor necrosis factor-α and Notch.30 TGF-β is an important molecule for the induction of EMT during metastasis and embryogenesis.31, 32, 33 TGF-β was shown to have two actions: tumor suppression and tumor promotion. TGF-β acts as a tumor repressor during early tumor growth and induces cell growth arrest and apoptosis. During late tumor growth, TGF-β initiates cancer progression and metastasis through smad-dependent or smad-independent signaling pathways.21, 34 For example, the exposure of mouse keratinocytes overexpressing TGF-β to chemical carcinogens results in the TGF-β-mediated suppression of skin tumor formation during the initial stages of cancer progression, while TGF-β acts as a tumor promoter during the late stages of cancer progression.35 The Smad-dependent signaling pathway regulates EMT through the expression of snail, ZEB and twist.20 These transcriptional regulators repress the expression of epithelial markers, such as E-cadherin, plakoglobin and occludin, and also activate mesenchymal markers, including vimentin, fibronectin and N-cadherin.20, 36 TGF-β signaling involves ligand binding to TGF receptors (TβR1 and TβR2). Smad2/3 is subsequently activated and forms a complex with smad4. Smad 2/3/4 complexes translocate to the nucleus and interact with other transcription factors to regulate the expression of target genes.37 Smad-independent pathways include phosphoinositide 3-kinase/Akt, Ras, mitogen-activated protein kinase and Rho-like GTPase pathways.38 These pathways upregulate the expression of snail, twist and ZEB. TGF-β also promotes EMT through interactions with Wnt and Notch.20
The Notch pathway is initiated through interactions between the Notch receptor and ligands on adjacent cells. Four Notch receptors (1–4) and five ligands (Dll-1, Dll-3, Dll-4, Jagged-1 and Jagged-2) exist in mammals.39, 40 Notch signaling is initiated through ligand binding to an adjacent receptor. Subsequently, γ-secretase cleaves the intramembrane Notch receptor. The released Notch intracellular domain translocates to the nucleus and interacts with C-protein-binding factor 1/Suppressor of Hairless/Lag-140, 41 and acts as an activator of target genes, including Hes and Hey. Although studies have suggested that Notch signaling is insufficient to completely induce EMT and crosstalk with other signaling molecules might therefore be required,21 the Notch signaling pathway has been considered an important regulator for the induction of EMT. Notch activation morphologically, phenotypically and functionally converts epithelial cells into mesenchymal-type cells.42 This change has been associated with the downregulation of epithelial markers, including E-cadherin, Tei1, Tei2 and platelet–endothelial epithelial cell adhesion molecule-1, and the upregulation of mesenchymal markers, such as α-smooth muscle actin (SMA) and fibronectin.42 It has been reported that snail and slug are induced through Notch signaling to promote EMT, which has an important role during both embryonic development and tumor progression.43, 44
Wnt signaling is also important for diverse cell functions via canonical (β-catenin) or noncanonical pathways.21 The formation of the Wnt–Fz–LRP complex through the binding of ligands, that is, wnt1 and wnt3, to their receptors, Frizzled (Fz) and LRP 5/6, initiates the canonical pathway. Without the Wnt signaling pathway, cytoplasmic β-catenin forms a complex with Axin, adenomatous polyposis coli, glycogen synthase kinase-3β and Ck1.45 When the cell receives Wnt signals, a complex is formed between LRP5/6 and Fz. These structures affect β-catenin stabilization, nuclear translocation and protein accumulation. In the nucleus, β-catenin forms a complex with T-cell factor/lymphoid enhancer factor, thereby initiating the expression of Wnt target genes.45, 46 During EMT, smad2 and smad 4 influence Wnt signaling to repress E-cadherin expression in medial-edge epithelial cells. Lymphoid enhancer factor-1 has also been associated with mesenchymal marker expression.20, 47 Other studies have suggested that wnt3 promotes EMT through the increased expression of N-cadherin, twist and slug and the decreased expression of E-cadherin in the trastuzumab-insensitive cells, SKBR3/100-8 and BT474/100-2.48 In addition, previous studies have shown that the activation of Wnt signaling is important for the induction of EMT in breast and prostate cancers.49, 50
RTK signaling alone does not induce EMT. Therefore, interplay with other signaling molecules might be required.21 Growth factors, such as hepatocyte growth factor, epidermal growth factor and fibroblast growth factor, bind to their respective receptors and activate extensive crosstalk networks that affect EMT.21 These growth factors switch signals through the structural activation of RTK, which activates TGF-β and integrin signaling pathways to alter EMT. Three activities occurring downstream of Ras might be required for the induction of EMT: smad2 activation, phosphoinositide 3-kinase/Akt pathway initiation and Raf/mitogen-activated protein kinase signaling pathway.30
EMT in ESCs
Few studies examining the role(s) of EMT in human and mouse ESCs have been published.51, 52 In most ESCs, the transformation of epithelial cells into mesenchymal cells might occur during ESC differentiation. In monolayer cultures, differentiation of human ESCs involves (1) the conversion from E-cadherin to N-cadherin, (2) increased vimentin expression, (3) the increase of repression molecules of E-cadherin, such as Snail and Slug, and (4) increased gelatinase activity and cellular motility. In undifferentiated human ESCs, the abrogation of E-cadherin-mediated cell–cell contact increases cellular motility and induces actin cytoskeleton rearrangement.53 The 5T4 antigen is expressed on the cell surface, generating a mesenchymal phenotype. The expression of the 5T4 oncofetal antigen is upregulated in colorectal, gastric and ovarian carcinomas.53, 54, 55, 56 Overexpression of 5T4 antigen on mouse ESCs reduces cell–cell contact, downregulates E-cadherin expression and alters the actin cytoskeleton in epithelial cells.53, 57, 58 The lack of 5T4 antigen is involved in increase of the restoration of cell–cell contact, and the upregulation of E-cadherin production prevents the development of a mesenchymal morphology.53 This effect might reflect the stabilization of the cortical actin cytoskeleton rearrangement through E-cadherin, which blocks 5T4 antigen localization. Human ESC colonies, cultured under feeder-free conditions, undergo differentiation and develop a mesenchymal cell-like phenotype.52 The mesenchymal-like cells express more mesenchymal markers compared with a single upper layer of columnar (zone 1) cells.52 In another study,59 EMT occurred not only in the embryoid body formed from human ESCs or human induced pluripotent stem cells but also in cultures of human pluripotent stem cells. Thus, it is reasonable to expect that human pluripotent stem cells resemble cells obtained from the epiblast. Thus, the occurrence of EMT in human pluripotent stem cells and embryoid bodies reflects the EMT observed during gastrulation in human development. In addition, the relationship between protein tyrosine kinase 7 (ptk7) and EMT was also examined in this study. The ptk7 has been considered an important factor for embryonic development in mice, particularly during gastrulation and neural tube closure.59, 60 Although ptk7-positive cells lose pluripotent marker and E-cadherin expression and acquire mesenchymal marker expression, ptk7 did not affect pluripotency or lineage marker changes.59 In mouse ESCs, Ell3, a testis-specific RNA polymerase 2 elongation factor, promotes lineage differentiation through the upregulation of EMT-associated gene expression and the downregulation of pro-apoptotic gene expression. The local expression of Ell3 has been observed in undifferentiated cells, and this expression affects embryoid body formation.61 However, Ell3 does not influence the expression of self-renewal markers, suggesting that ZEB1 might have an important role in differentiation through Ell3. Thus, EMT in ESCs might induce differentiation through several pathways.
MicroRNA acts as a regulator of EMT in many cell types. For example, snail promotes EMT through the repression of microRNA 200 family members and induces mesoderm differentiation during the epiblast stem cell stage, suggesting that microRNA 200 family members might suppress EMT and the differentiation of ESCs at the epiblast stem cell stage.62 Another study reported that the mesenchymal–epithelial transition might be important in reprogramming fibroblasts. OCT4, SOX2, Klf4 and c-Myc are imported into fibroblasts, and these proteins induce the rapid downregulation of microRNA-155 and microRNA-10b, associated with EMT, and the upregulation of microRNA-205 and -429 during the development of induced pluripotent stem cells.63 Therefore, it is reasonable to assume that EMT might be associated with the differentiation of ESCs, suggesting that certain factors, that is, the microRNA family, which regulate EMT at various pluripotent stages, might exist. Naïve cells are thought to be more undifferentiated than primed cells; if this is correct, then these regulating factors may have an influence on pluripotent stages of naïve and primed cells.
Conclusion
EMT is important for embryonic development, and this process affects metastasis and the invasion of various cancers. Several molecules, including TGF-β and other growth factors, induce EMT. These factors bind to their respective receptors and might also interact with each other. As shown in Figure 1, various signaling pathways (for example, the TGF-β, Wnt/glycogen synthase kinase-3β, Notch and RTK signaling pathways) might be critical for the induction of EMT. ESCs are pluripotent, suggesting that these cells can differentiate into many cell types (Figure 2). Although the exact mechanism(s) underlying EMT activity has not been clarified in these cells, ESCs might be controlled through EMT under various circumstances during differentiation. Transcriptional activators of EMT, such as snail and ZEB, have also been implicated in the differentiation of ESCs, and regulating factors, such as the microRNA family, specifically promoted or inhibited EMT at the pluripotent cell stage. Thus, ESCs remain at either the pluripotent stage or the more differentiated stage. EMT and regulating factors regulate the differentiation of ESCs. Because ESCs, rather than epiblast stem cells, are more undifferentiated, these cells undergo EMT differentiation, suggesting that this process contributes to naïve and primed cell types. Thus, further studies should examine the mechanism(s) underlying EMT in ESCs required to generate transgenic organisms and induced pluripotent stem cells in order to develop the treatments for diseases using stem cells.

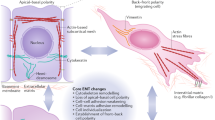
Signaling pathways that induce epithelial–mesenchymal transition (EMT). Transforming growth factor (TGF)-β might affect both Smad-dependent and -independent pathways. Smad2 and Wnt activate the Notch signaling pathway. The Notch receptor is cleaved, and subsequently the Notch intracellular domain (NIC) translocates into the nucleus. The role of this protein shifts from that of a repressor to an activator through binding to DNA-binding proteins, such as CBF1, Su (H) and LAG-1. Wnt signaling is initiated through the inhibition of glycogen synthase kinase (GSK)-3β. β-Catenin subsequently enters the nucleus and forms a complex with lymphoid enhancer factor/T-cell factor to control the expression of target genes. Receptor tyrosine kinase (RTK) has a critical role in the induction of these intermediates. PI3K, phosphoinositide 3-kinase.

Potential effect of epithelial–mesenchymal transition (EMT) on the differentiation of embryonic stem cells (ESCs). Transcription factors, that is, the snail family, and regulatory factors, that is, microRNA, that regulate downstream signaling pathways associated with the EMT response might affect EMT in ESCs. ESCs (embryo blastocyst; undifferentiated) differentiate into many cell types in the body, suggesting that EMT might be involved in the differentiation of ESCs.
References
Thiery JP, Acloque H, Huang RY, Nieto MA . Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139: 871–890.
Polyak K, Weinberg RA . Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009; 9: 265–273.
Kalluri R, Weinberg RA . The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119: 1420–1428.
Sohal SS, Walters EH . Role of epithelial mesenchymal transition (EMT) in chronic obstructive pulmonary disease (COPD). Respir Res 2013; 14: 120.
Rippon HJ, Bishop AE . Embryonic stem cells. Cell Prolif 2004; 37: 23–34.
Evans MJ, Kaufman MH . Establishment in culture of pluripotential cells from mouse embryos. Nature 1981; 292: 154–156.
Ginis I, Luo Y, Miura T, Thies S, Brandenberger R, Gerecht-Nir S et al. Differences between human and mouse embryonic stem cells. Dev Biol 2004; 269: 360–380.
Hanna J, Cheng AW, Saha K, Kim J, Lengner CJ, Soldner F et al. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc Natl Acad Sci USA 2010; 107: 9222–9227.
Nichols J, Smith A . Naive and primed pluripotent states. Cell Stem Cell 2009; 4: 487–492.
Park JK, Kim HS, Uh KJ, Choi KH, Kim HM, Lee T et al. Primed pluripotent cell lines derived from various embryonic origins and somatic cells in pig. PLoS ONE 2013; 8: e52481.
Mohamet L, Hawkins K, Ward CM . Loss of function of e-cadherin in embryonic stem cells and the relevance to models of tumorigenesis. J Oncol 2011; 2011: 352616.
Pertz O, Bozic D, Koch AW, Fauser C, Brancaccio A, Engel J . A new crystal structure, Ca2+ dependence and mutational analysis reveal molecular details of E-cadherin homoassociation. EMBO J 1999; 18: 1738–1747.
Perez-Moreno M, Fuchs E . Catenins: keeping cells from getting their signals crossed. Dev Cell 2006; 11: 601–612.
Xiong H, Hong J, Du W, Lin YW, Ren LL, Wang YC et al. Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J Biol Chem 2012; 287: 5819–5832.
Di Croce L, Pelicci PG . Tumour-associated hypermethylation: silencing E-cadherin expression enhances invasion and metastasis. Eur J Cancer 2003; 39: 413–414.
Nieto MA . The snail superfamily of zinc-finger transcription factors. Nat Rev 2002; 3: 155–166.
Sanchez-Tillo E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M et al. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci 2012; 69: 3429–3456.
Vandewalle C, Van Roy F, Berx G . The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci 2009; 66: 773–787.
Xue G, Hemmings BA . Phosphorylation of basic helix-loop-helix transcription factor Twist in development and disease. Biochem Soc Trans 2012; 40: 90–93.
Xu J, Lamouille S, Derynck R . TGF-beta-induced epithelial to mesenchymal transition. Cell Res 2009; 19: 156–172.
Garg M . Epithelial-mesenchymal transition—activating transcription factors—multifunctional regulators in cancer. World J Stem Cells 2013; 5: 188–195.
Jorda M, Olmeda D, Vinyals A, Valero E, Cubillo E, Llorens A et al. Upregulation of MMP-9 in MDCK epithelial cell line in response to expression of the Snail transcription factor. J Cell Sci 2005; 118: 3371–3385.
Taki M, Verschueren K, Yokoyama K, Nagayama M, Kamata N . Involvement of Ets-1 transcription factor in inducing matrix metalloproteinase-2 expression by epithelial-mesenchymal transition in human squamous carcinoma cells. Int J Oncol 2006; 28: 487–496.
Medici D, Hay ED, Olsen BR . Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell 2008; 19: 4875–4887.
Peinado H, Olmeda D, Cano A . Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 2007; 7: 415–428.
Khan MA, Chen HC, Zhang D, Fu J . Twist: a molecular target in cancer therapeutics. Tumour Biol 2013; 34: 2497–2506.
Fu J, Qin L, He T, Qin J, Hong J, Wong J et al. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res 2011; 21: 275–289.
Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 2008; 9: 582–589.
Das S, Becker BN, Hoffmann FM, Mertz JE . Reversal of transforming growth factor-beta induced epithelial-to-mesenchymal transition and the ZEB proteins. Fibrogenesis Tissue Repair 2012; 5 (Suppl 1): S28.
Thiery JP . Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol 2003; 15: 740–746.
Lee J, Choi JH, Joo CK . TGF-beta1 regulates cell fate during epithelial-mesenchymal transition by upregulating survivin. Cell Death Dis 2013; 4: e714.
Miyazono K . Transforming growth factor-beta signaling in epithelial-mesenchymal transition and progression of cancer. Proc Jpn Acad Ser B Phys Biol Sci 2009; 85: 314–323.
Yang Y, Pan X, Lei W, Wang J, Song J . Transforming growth factor-beta1 induces epithelial-to-mesenchymal transition and apoptosis via a cell cycle-dependent mechanism. Oncogene 2006; 25: 7235–7244.
Derynck R, Zhang YE . Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003; 425: 577–584.
Elliott RL, Blobe GC . Role of transforming growth factor Beta in human cancer. J Clin Oncol 2005; 23: 2078–2093.
Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117: 927–939.
Grady WM . Transforming growth factor-beta, Smads, and cancer. Clin Cancer Res 2005; 11: 3151–3154.
Zhang YE . Non-Smad pathways in TGF-beta signaling. Cell Res 2009; 19: 128–139.
Miele L . Notch signaling. Clin Cancer Res 2006; 12: 1074–1079.
Wang Z, Li Y, Kong D, Sarkar FH . The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr Drug Targets 2010; 11: 745–751.
Bray SJ . Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 2006; 7: 678–689.
Noseda M, McLean G, Niessen K, Chang L, Pollet I, Montpetit R et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ Res 2004; 94: 910–917.
Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A . Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol 2008; 182: 315–325.
Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev 2004; 18: 99–115.
MacDonald BT, Tamai K, He X . Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009; 17: 9–26.
Rao TP, Kuhl M . An updated overview on Wnt signaling pathways: a prelude for more. Cir Res 2010; 106: 1798–1806.
Nawshad A, Medici D, Liu CC, Hay ED . TGFbeta3 inhibits E-cadherin gene expression in palate medial-edge epithelial cells through a Smad2-Smad4-LEF1 transcription complex. J Cell Sci 2007; 120: 1646–1653.
Wu Y, Ginther C, Kim J, Mosher N, Chung S, Slamon D et al. Expression of Wnt3 activates Wnt/beta-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol Cancer Res 2012; 10: 1597–1606.
Jiang YG, Luo Y, He DL, Li X, Zhang LL, Peng T et al. Role of Wnt/beta-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1alpha. Int J Urol 2007; 14: 1034–1039.
Wu ZQ, Li XY, Hu CY, Ford M, Kleer CG, Weiss SJ . Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc Natl Acad Sci USA 2012; 109: 16654–16659.
Ullmann U, Gilles C, De Rycke M, Van de Velde H, Sermon K, Liebaers I . GSK-3-specific inhibitor-supplemented hESC medium prevents the epithelial-mesenchymal transition process and the up-regulation of matrix metalloproteinases in hESCs cultured in feeder-free conditions. Mol Hum Reprod 2008; 14: 169–179.
Ullmann U, In't Veld P, Gilles C, Sermon K, De Rycke M, Van de Velde H et al. Epithelial-mesenchymal transition process in human embryonic stem cells cultured in feeder-free conditions. Mol Hum Reprod 2007; 13: 21–32.
Eastham AM, Spencer H, Soncin F, Ritson S, Merry CL, Stern PL et al. Epithelial-mesenchymal transition events during human embryonic stem cell differentiation. Cancer Res 2007; 67: 11254–11262.
Naganuma H, Kono K, Mori Y, Takayoshi S, Stern PL, Tasaka K et al. Oncofetal antigen 5T4 expression as a prognostic factor in patients with gastric cancer. Anticancer Res 2002; 22: 1033–1038.
Starzynska T, Rahi V, Stern PL . The expression of 5T4 antigen in colorectal and gastric carcinoma. Br J Cancer 1992; 66: 867–869.
Wrigley E, McGown AT, Rennison J, Swindell R, Crowther D, Starzynska T et al. 5T4 oncofetal antigen expression in ovarian carcinoma. Int J Gynecol Cancer 1995; 5: 269–274.
Carsberg CJ, Myers KA, Stern PL . Metastasis-associated 5T4 antigen disrupts cell-cell contacts and induces cellular motility in epithelial cells. Int J Cancer 1996; 68: 84–92.
Ward CM, Barrow K, Woods AM, Stern PL . The 5T4 oncofoetal antigen is an early differentiation marker of mouse ES cells and its absence is a useful means to assess pluripotency. J Cell Sci 2003; 116: 4533–4542.
Chan DN, Azghadi SF, Feng J, Lowry WE . PTK7 marks the first human developmental EMT in vitro. PLoS ONE 2012; 7: e50432.
Yen WW, Williams M, Periasamy A, Conaway M, Burdsal C, Keller R et al. PTK7 is essential for polarized cell motility and convergent extension during mouse gastrulation. Development (Cambridge, England) 2009; 136: 2039–2048.
Ahn HJ, Cha Y, Moon SH, Jung JE, Park KS . Ell3 enhances differentiation of mouse embryonic stem cells by regulating epithelial-mesenchymal transition and apoptosis. PLoS ONE 2012; 7: e40293.
Gill JG, Langer EM, Lindsley RC, Cai M, Murphy TL, Kyba M et al. Snail and the microRNA-200 family act in opposition to regulate epithelial-to-mesenchymal transition and germ layer fate restriction in differentiating ESCs. Stem Cells (Dayton, Ohio) 2011; 29: 764–776.
Li R, Liang J, Ni S, Zhou T, Qing X, Li H et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010; 7: 51–63.
Acknowledgements
This work was supported by a grant from the Next-Generation BioGreen 21 Program (no. PJ009599), Rural Development Administration, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Kim, YS., Yi, BR., Kim, NH. et al. Role of the epithelial–mesenchymal transition and its effects on embryonic stem cells. Exp Mol Med 46, e108 (2014). https://doi.org/10.1038/emm.2014.44
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2014.44
This article is cited by
-
FoxM1 contributes to progestin resistance and epithelial-to-mesenchymal transition in endometrial carcinoma
Molecular & Cellular Toxicology (2023)
-
Increased expression of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase-3 is required for growth of mouse embryonic stem cells that are undergoing differentiation
Cytotechnology (2023)
-
Acetylation stabilizes stathmin1 and promotes its activity contributing to gallbladder cancer metastasis
Cell Death Discovery (2022)
-
Rg3 and Rh2 ginsenosides suppress embryoid body formation by inhibiting the epithelial-mesenchymal transition
Archives of Pharmacal Research (2022)
-
Implications of reactive oxygen species in lung cancer and exploiting it for therapeutic interventions
Medical Oncology (2022)
