
Abstract
Apurinic/apyrimidinic endonuclease 1 (APE1) is a multifunctional enzyme involved in the base excision repair (BER) pathway, which repairs oxidative base damage caused by endogenous and exogenous agents. APE1 acts as a reductive activator of many transcription factors (TFs) and has also been named redox effector factor 1, Ref-1. For example, APE1 activates activator protein-1, nuclear factor kappa B, hypoxia-inducible factor 1α, paired box gene 8, signal transducer activator of transcription 3 and p53, which are involved in apoptosis, inflammation, angiogenesis and survival pathways. APE1/Ref-1 maintains cellular homeostasis (redox) via the activation of TFs that regulate various physiological processes and that crosstalk with redox balancing agents (for example, thioredoxin, catalase and superoxide dismutase) by controlling levels of reactive oxygen and nitrogen species. The efficiency of APE1/Ref-1’s function(s) depends on pairwise interaction with participant protein(s), the functions regulated by APE1/Ref-1 include the BER pathway, TFs, energy metabolism, cytoskeletal elements and stress-dependent responses. Thus, APE1/Ref-1 acts as a ‘hub-protein’ that controls pathways that are important for cell survival. In this review, we will discuss APE1/Ref-1’s versatile nature in various human etiologies, including neurodegeneration, cancer, cardiovascular and other diseases that have been linked with alterations in the expression, subcellular localization and activities of APE/Ref-1. APE1/Ref-1 can be targeted for therapeutic intervention using natural plant products that modulate the expression and functions of APE1/Ref-1. In addition, studies focusing on translational applications based on APE1/Ref-1-mediated therapeutic interventions are discussed.
Similar content being viewed by others
Introduction
A number of human pathologies, including cancer, neurodegenerative diseases (NDs) and cardiovascular disorders (CVDs), result from oxidative DNA damage caused by endogenous and exogenous agents,1, 2, 3 as summarized in Figure 1. The base excision repair (BER) pathway is the main repair pathway that repairs base damage (alkylation, oxidization, uracil, apurinic/apyrimidinic (AP) sites and strand breaks) in both the cell nucleus and mitochondria.4, 5 AP site formation is central to this process and occurs spontaneously due to the attack of reactive oxygen species (ROS) or specifically due to the action of DNA glycosylase (DG).6 AP endonuclease 1 (APE1), a multifunctional enzyme that is part of the BER pathway, exhibits DNA repair activity and has a role in the reductive activation of many transcription factors (TFs).7 These two activities are encoded by two distinct regions of the APE1 protein: the N-terminal region encodes the redox function and the C-terminal region encodes the repair function.8, 9 The DNA repair activity involves AP endonuclease activity, 3′ phosphodiesterase activity, 3′ phosphatase activity, and 3′–5′ exonuclease activity7, 10 and engages in protein–protein interactions with BER pathway proteins.6, 11 APE1 physically interacts with BER pathway participant proteins, such as poly (ADP-ribose) polymerase 1 (PARP1),12 X-ray repair cross-complementing protein 1 (XRCC1),13 DNA polymerase β,14 flap endonuclease 1 (FEN-1) and proliferating cell nuclear antigen15 to stimulate individual proteins with which it interacts; thus, APE-1 coordinates the BER pathway.6

The role of apurinic/apyrimidinic endonuclease 1 (APE1)/redox effector factor-1 (Ref-1) in maintaining genome integrity in response to reactive oxygen species/reactive nitrogen species (ROS/RNS)-mediated oxidative stress associated with various human diseases and the possible intervention of phytochemicals toward the functional modulation of APE1/Ref-1. ROS/RNS from endogenous and exogenous sources cause oxidative stress in various cell types; in response to this stress, APE1/Ref-1 dual functions (repair and redox) are activated as a part of the DNA repair response through the base excision repair (BER) pathway to maintain the genomic integrity of the cell and the redox regulation of various transcription factors (TFs) in cell-survival pathways. Natural plant products hold the potential to modulate the expression and functions of APE1/Ref-1, thus representing a promising therapeutic intervention against various human diseases including cancer, cardiovascular and neurodegenerative disorders. EGCG, epigallocatechingallate; UV, ultraviolet.
APE1 has another important function, in which respect it is also known as the redox effector factor 1 (Ref-1).7 APE1/Ref-1 reductively activates TFs including c-Jun, activator protein-1 (AP-1), nuclear factor kappa B (NF-κB), the tumor-suppressor protein p53, hypoxia-inducible factor 1α (HIF-1α) and paired box gene 8, which are involved in various cellular processes such as cell survival, growth signaling and inflammatory pathways.16, 17, 18, 19, 20 The mechanism by which APE1/Ref-1 reduces the cysteine (Cys) residues of these TFs involves the exchange of a proton (H+ ion) from one or two of the redox active Cys residues in its N-terminus (Cys65, Cys93, Cys99 or Cys138), thereby activating their DNA-binding activity.7, 21
Specific redox-dependent and redox-independent physical protein–protein interactions are also mediated by APE1/Ref-1 to carry out other biologically relevant cellular functions such as that involving Werner (WRN) protein, which is involved in premature aging;22 ERp57 (endoplasmic reticulum-associated p57 (thiol-disulfide oxidoreductase)) protein, which is involved in the disulfide shuffling of glycoproteins;23 Y-box-binding protein 1 (YB-1), which causes the activation of the multidrug resistance (MDR1) gene;24, 25 and various neuronal proteins (such as pyruvate kinase 3 isoform 2 (PKM2), tropomodulin 3 (Tmod3) and heterogeneous nuclear ribonucleoprotein-H1 (hn-RNP-H)), which are involved in neuronal cell survival after amyloid beta (Aβ) (25–35) peptide-induced stress-dependent events.26 APE1/Ref-1 dysregulation has been associated with neurodegenerative27, 28, 29, 30 and CVDs31 and with various human cancers.32, 33, 34, 35, 36, 37 APE1/Ref-1 may have a role in cancer progression via its ability to increase DNA repair and anti-apoptotic, inflammatory and growth-promoting activities.21, 34 In NDs, decreases in the activities of APE1/Ref-1 contribute toward disease progression, and neuronal cell survival is aided by increasing or protecting APE1/Ref-1’s activities.26
DNA damage and repair can have a key role in cancer therapy-induced neurotoxicity; to establish that such a phenomenon exists, Englander’s group demonstrated the role of APE1/Ref-1 in chemotherapy-induced peripheral neuropathy (CIPN). Peripheral nervous system (PNS) neurons were shown to be vulnerable to circulating toxin/inflammatory mediator/anticancer agent-induced DNA damage due to the permeability of the PNS blood–nerve barrier.38 Thus, CIPN can obstruct anticancer treatment goals. This obstruction can be overcome by improving DNA repair fidelity in PNS neurons. Another study has observed the overexpression and localization of DNA repair proteins (including APE1/Ref-1) in antimitotic agent-induced damage to dorsal root ganglia (DRG) neurons and noted the potential for using various repair proteins for use as biomarkers and for the reversal of cancer therapy-induced damage; further, they noted that these proteins can be exploited for use in predicting post-treatment risks in PNS neurons.39
Despite the correct execution of the endonuclease and redox activity of APE1/Ref-1, alterations in the protein–protein interaction(s) between APE1/Ref-1 and other proteins of the BER or other DNA-transacting pathways can modulate the final consequences of such interactions. Thus, APE1/Ref-1’s role in regulating DNA repair, in the activation or repression of various TFs, and in maintaining the cellular redox state means that APE1/Ref-1 has a role in contributing to or preventing wide range of human pathologies.40 Thus, APE1/Ref-1 is special in terms of its expression pattern, subcellular localization, repair, redox activity and protein–protein interaction, indicating its therapeutic potential (Figure 2). In this review, we emphasize how natural plant products that modulate APE1/Ref-1’s expression and functions can be used as adjuvant in conjunction with standard therapies for various human diseases.

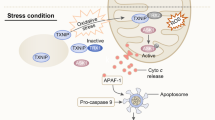
Stress-induced prismatic functions of apurinic/apyrimidinic endonuclease 1 (APE1)/redox effector factor-1 (Ref-1) in different cell types during disease conditions. Activation of APE1/Ref-1 occurs because of various external and internal stimulating factors (for example, radiation, reactive oxygen species/reactive nitrogen species (ROS/RNS) and hypoxia). In addition, many misfolded proteins, namely, amyloid beta (Aβ), α- synuclein, Tau and PrPsc, also cause APE1/Ref-1 activation. Further increases in [Ca2+]i also induce APE1/Ref-1 expression in various cell types. Upregulation of APE1/Ref-1 further activates various proteins either directly or indirectly and controls and modulates various key biological functions including DNA repair, cellular redox balance, cell proliferation, cytoskeleton modification and energy metabolism.
APE1/Ref-1: a multifunctional enzyme
APE1/Ref-1 is expressed ubiquitously in humans at a concentration of approximately 0.35 to 7 × 106 molecules per cell.41 This 36.5-kDa protein is encoded by a 3-kb gene, which is located on chromosome 14.42 The active site for the DNA repair function of APE1/Ref-1 is located within the C-terminal domain; three residues in this domain (Phe266, Trp280 and Leu282) form a hydrophobic pocket that recognizes and captures the AP site. APE1/Ref-1 is a rate-limiting enzyme in BER pathway; the efficiency of the fast step of incision at the AP site can be limited by slow product release.43 The enzyme works by first inserting its loop into both the major and minor grooves of DNA, then flipping the AP nucleotide into its hydrophobic pocket and finally kinking the DNA helix.44, 45 The N-terminus contains a nuclear localization signal that is crucial for redox activity.7, 45, 46, 47 APE1/Ref-1 has a role in distinct transcriptional regulatory functions by acting as a reductive activator of TFs and acting as a redox-independent transcriptional repressor of some genes.17, 18, 19, 20, 21 Oxidative damage to both DNA and RNA (including messenger RNA, ribosomal RNA and transfer RNA) have been associated with various diseases.48 Recently, APE1/Ref-1’s role in RNA metabolism (including its function as a riboendonuclease) has been identified, showing that it has a role in the post-transcriptional regulation of gene expression.49, 50 Therefore, APE1/Ref-1 is a multifunctional enzyme having repair, redox and RNA metabolism activities, indicating its potential for use as an anticancer and neuroprotective agent in the development of improved therapies.
The role of APE1/Ref-1 in oxidative DNA damage repair
Oxidatively damaged DNA accelerates the development of diseases including cancer and NDs involving alterations in gene sequences and signaling pathways. Almost all oxidative DNA base lesions are repaired via the BER pathway.51 The formation of an intermediate AP site during the processing and repair of oxidatively damaged DNA bases via the BER pathway is common when the removal of DNA base lesion is mediated either spontaneously by ROS or through the action of monofunctional DGs during single-nucleotide (SN)-BER.6, 52 APE1/Ref-1, the second enzyme in the BER pathway (Figure 3), then hydrolyzes the phosphodiester backbone immediately 5′ to the generated AP site to produce a 3′ OH group and a 5′ deoxyribose-5-phosphate blocking group.53, 54 In long-patch (LP)-BER, a bifunctional DG with additional AP lyase activity generates 3′ phospho α, β-unsaturated aldehyde and 3′ phosphate termini via β elimination reaction at the site of strand cleavage. The 3′ phosphodiesterase activity of APE1/Ref-1 helps to remove the β-unsaturated aldehyde and 3′ termini phosphate.6, 52 LP-BER utilizes polymerase delta (pol δ) and epsilon (pol ɛ), FEN-1 and proliferating cell nuclear antigen, and Lig I.6, 52 The type of lesion formed and the initial levels of initiating DG and cellular ATP determine the level of involvement of the BER sub-pathway.6, 11, 52 SN-BER and LP-BER both require the generation of 3′OH termini by APE1/Ref-1 but utilize different enzymes for subsequent completion of DNA repair.6, 52 Hence, APE1/Ref-1 is an indispensable safeguard against oxidative DNA damage.55 Following removal of the blocking group (dRP) by the dRP lyase activity of DNA pol β, DNA repair is completed by the action of DNA ligase, thus restoring genome integrity.56 The bifunctional DGs with additional intrinsic AP lyase activity cleave the DNA strand 3′ to the AP site, thereby generating a 3′ blocking group.57, 58 The resulting 3′ blocking group is then removed by the APE1/Ref-1 enzyme (or in some cases, by polynucleotide kinase).41, 59 The 3′ phosphodiesterase activity of APE1/Ref-1 is also involved in repairing single-strand breaks in DNA, and a 3′ blocking group is directly generated by ROS.60 Unrepaired AP sites can lead to DNA strand breaks, apoptosis and increased cytotoxicity.61 Therefore, the DNA repair function of APE1/Ref-1 is indispensable in protecting the cell from both endogenous and exogenous DNA damage. All APEs possess both endonuclease and 3′ phosphodiesterase activity.53, 54 However, the mammalian endonuclease activity of APE1/Ref-1 is stronger than its 3′ exonuclease/phosphodiesterase activity.41, 53 Furthermore, unlike Escherichia coli exonuclease III (xthA), which exhibits potent 3′ phosphatase/phosphodiesterase activities, mammalian APE1/Ref-1 exhibits extremely weak 3′ phosphatase activity, which is required to remove 3′ phosphate groups that are directly generated by ROS or that result from the AP lyase activity of mammalian DGs, NEIL1 (endonuclease VIII-like 1) and NEIL2.62, 63

A schematic representation of the mechanism of the short-patch (SN-BER) and long-patch (LP-BER) sub-pathways of base excision repair (BER) in mammalian cells. apurinic/apyrimidinic endonuclease 1 (APE1)/redox effector factor-1 (Ref-1) functions as an AP endonuclease in SN-BER, initiated by monofunctional DNA glycosylase (DG), and as a 3′phosphodiesterase in LP-BER, initiated by bifunctional DG. Pol β, X-ray repair cross-complementing protein 1 (XRCC1) and ligase III are required for SN-BER to conduct SN repair, whereas proliferating cell nuclear antigen (PCNA), Pol δ/ɛ, flap endonuclease 1 (FEN-1) and ligase I are required for LP-BER to conduct multinucleotide repair in mammalian cells.
The role of APE1/Ref-1 in redox regulation
Redox potential controls gene expression by regulating the DNA-binding activity of TFs.7 APE1 is also known as Ref-1 because of its role in the redox regulation of the DNA-binding activity of various TFs.7 The ability of Ref-1 to facilitate the DNA-binding activity of AP-1 was identified in HeLa cells.20 A thiol exchange reaction was found to be involved in the reductive activation of c-Jun by reduced APE1/Ref-1, in which the Cys272 of c-Jun is first oxidized to sulfenic acid (SOH) and then reduced by Cys65 of APE1/Ref-1 to the active form.20 The resultant oxidized form of APE1/Ref-1 is then reduced by thioredoxin (TRX), and oxidized TRX is reduced by TRX reductase.64 Seven Cys residues have been reported as conserved in mammalian APE1. Of the seven Cys residues, three (Cys65, Cys93 and Cys99) are considered important for redox function.65 Cys65 and Cys93 are buried and surface inaccessible, whereas Cys99 is solvent accessible. A study by Walker et al.66 reported that Cys65 is essential for redox function in vitro. In this study, each of seven Cys residues was substituted by Ala singly, and each mutant was then tested for redox activity. Only Cys65Ala lost redox activity. Cys93Ala retained redox activity but at significantly lower levels than in wild-type (WT) APE1. A possible explanation for the redox-inactivity of the Cys65Ala mutant may be a change in the stability and folding of APE1.67 Nuclear magnetic resonance (NMR) evidence demonstrated that TRX interacts with a Cys65-containing polypeptide at the active site.68 A study showed that the Cys65 mutant acts as a dominant-negative inhibitor of the redox function of selenomethionine-containing APE1/Ref-1.69 Furthermore, the apoptosis of neurons after oxidative stress has been shown to be prevented by ectopic WT APE1/Ref-1 expression and also, but to a smaller extent, by the Cys65Ala mutant.70 Moreover, several studies using TF-dependent reporter expression assays have shown that the Cys65 mutant does not behave like WT APE1/Ref-1.71, 72, 73 However, the proposed Cys65-mediated redox function of APE1/Ref-1 was challenged by Curran’s group,74 which showed that homozygous Cys64Ala (in humans, Cys65) knock-in mouse APE1/Ref-1 mutants are viable and retain normal levels of AP-1 DNA-binding activity. Furthermore, the recombinant Cys64Ala mutant exhibited normal c-Jun reducing activity, similar to that exhibited by WT APE1/Ref-1 in vitro.74 Another study showed that APE1/Ref-1 stimulates p50 or c-Jun DNA-binding by facilitating their reduction by reducing agents such as glutathione or TRX, and none of the Cys residues in APE1/Ref-1 were found to be essential for this activity.75 These findings challenge the role of Cys64/65 in the redox function of APE1/Ref-1. However, these findings have been disproved by some recent studies. Luo et al.76 discovered that Thr58 is equivalent to Cys65 in zebrafish. It was observed that substituting Thr58 for Cys in the zebrafish enzyme confers a redox function, as found using in vitro assays and transactivation assays.77 Another study by Vascotto et al.78 has also indicated that Cys65 has a role in controlling APE1/Ref-1 sub-cellular functions. Double-cysteine mutants have been also analyzed to determine whether other Cys residues have roles in the redox activity. All double-cysteine mutants except Cys93Ala/Cys99Ala displayed redox activity.65 This study further suggested that other Cys residue(s) of APE1 may have a role in the redox mechanism.
Specific redox inhibitors and mass spectral (MS) analysis have also been used to characterize the detailed mechanism and redox active form of APE1/Ref-1. In this context, E3330 has been used in various studies, and the mechanism of interaction between E3330 and APE1/Ref-1 has remained a subject of interest.77, 79 In one of the studies by Zhang et al.80 using MS and N-ethylmaleimide (NEM) labeling, it was concluded that E3330 interacts with partially unfolded APE1/Ref-1. In chemical foot-printing experiments with NEM, the incubation of APE1 with E3330 yielded fully modified APE1/Ref-1 protein; that is, all seven Cys residues were labeled with NEM. As previously mentioned, only two of the seven Cys residues are solvent accessible, therefore, APE/Ref-1 may be unfolded in the presence of E3330. Cys65, which is normally buried and is critical for the redox function of APE1/Ref-1, also becomes exposed and reacts with NEM in the presence of E3330. Furthermore, based on experiments using tandem MS and liquid chromatography, the E3330-mediated increase in disulfide bond formation of the critical Cys residues Cys65 and Cys93 of APE1/Ref-1 has been suggested as the cause of the inhibition of redox activity.81
The transcriptional activity of AP-1 is regulated by the direct interaction of APE1/Ref-1 and TRX.82 APE1/Ref-1 has been identified as capable of inducing the DNA-binding activity of NF-κB, a major TF that is involved in immune and inflammatory signaling pathways, and it has been found that loss of APE1/Ref-1 decreases NF-κB activity in endothelial cells, thereby increasing cellular susceptibility to apoptosis.83 APE1/Ref-1 has also been shown to regulate the activation of HIF-1α and p53 through its redox activity. The activation of the tumor-suppressor protein p53 by APE1/Ref-1 contributes to the response to oxidative stress. Activated p53 then migrates to the nucleus to further stimulate the transcription of p53-responsive genes, such as p21, cyclin G and BCL2-associated X protein (BAX).84 In addition, it has been demonstrated that APE1/Ref-1 controls paired box gene 8 DNA-binding activity, which is important for thyroid gland development.19 APE1/Ref-1 acts as a transcriptional coactivator and a corepressor either directly or indirectly by redox-dependent or redox-independent pathways. APE1/Ref-1 regulation occurs at both transcriptional and post-translational levels. At the transcriptional level, stimuli such as hormones and ROS increase the protein synthesis and nuclear translocation of APE1/Ref-1.85, 86, 87, 88
APE1/Ref-1 is regulated at the post-translational level via post-translational modifications such as acetylation, phosphorylation and ubiquitination.7, 11, 47 These modifications also regulate protein stability, interaction and intracellular distribution.89 APE1/Ref-1 is acetylated at Lys6 or Lys7, Lys25, Lys27 and Lys31.7, 90 The acetylation of Lys6 and Lys7 strongly affects the transcriptional regulatory functions of APE1/Ref-1.7 APE1/Ref-1 stably interacts with YB-1, and acetylation further enhances its binding with YB-1 both in vivo and in vitro,24 leading to activation of the MDR1 gene. It appears that an acetylation-mediated conformational change occurs in the disordered N-terminal segment (∼40 residues) of APE1/Ref-1 and that this conformational change is required for endonuclease activity and the modulation of protein–protein interactions.24, 25, 91, 92
Potential phosphorylation sites on APE1/Ref-1 include consensus sequences for casein kinase I/casein kinase II (CKI and CKII) and protein kinase C (PKC). PKC phosphorylation has been shown to stimulate the redox activity of APE1/Ref-1 in vitro.85, 93 Moreover, CKII-mediated phosphorylation abolished DNA repair activity in vitro, whereas phosphorylation by CKI or PKC had no such effect.94 Further, the phosphorylation of APE1/Ref-1 by CKII also enhanced the redox activation of the TF, AP-1.95 APE1/Ref-1 was found to be phosphorylated at Thr233 by the threonine kinase cyclin-dependent kinase 5 (CDK5), a paralog of CDK2/4.28 A high level of phosphorylated Thr233 was observed in brain tissues from PD and AD patients.28 It is possible that the inactivation and degradation of APE1/Ref-1 by phosphorylation and subsequent ubiquitination may profoundly affect the severity of various human diseases.
Ubiquitination has been demonstrated to have a role in regulating the steady-state levels of BER enzymes in mammalian cells by directing their degradation and turnover.96 Ubiquitin is a highly conserved small protein (∼8.5 kDa), the covalent addition of which to a target protein as a polymer by ubiquitin ligases promotes the latter’s degradation by the 26S proteasome.97 However, ubiquitinated proteins may also be recycled by the removal of ubiquitin by deubiquitinases or by ubiquitin-specific proteases.98, 99 Proteins may be ubiquitinated with a single ubiquitin moiety (monoubiquitination) or with multiple ubiquitin molecules (polyubiquitination). Recently, it has been demonstrated that APE1/Ref-1 ubiquitination, which is enhanced by phosphorylation at Thr233,100 can act as a signal to regulate the stability, subcellular localization and gene regulatory functions of APE1/Ref-1.93, 98
The specific interaction(s) between APE1 and TRX require the translocation of TRX from the cytoplasm to the nucleus,101 which can be induced by oxidants, cytokines and ultraviolet irradiation.102, 103 The results of one study indicate that APE1/Ref-1 promotes cell proliferation by acting as a direct redox regulator of extracellular signal-regulated kinase.104 Together, these studies suggest that APE1/Ref-1 is an essential enzyme that is responsible for the redox regulation of various TFs, either directly or indirectly, while involving other reducing factors.
The role of APE1/Ref-1 in maintaining cellular homeostasis
Cellular homeostasis is maintained through the balancing of cellular redox states. Any imbalance causes oxidative stress in cells. Cellular redox state is regulated by ROS/reactive nitrogen species (RNS) and enzymatic/non-enzymatic antioxidant systems.105, 106 ROS/RNS have significant roles in maintaining cellular redox states because they are continuously generated in cells during normal metabolism as respiration by-products and during inflammatory responses.107, 108, 109 Physiologically, low basal levels of ROS/RNS are involved in intracellular signaling as second messengers, whereas high levels of ROS/RNS cause oxygen and nitrogen toxicity.107, 110 ROS can also be induced exogenously by environmental agents such as heavy metals, ionizing radiation and chemotherapeutic drugs.111, 112 RNS, such as nitric oxide (NO·), are generated by NO synthase (NOS), endothelial NOS (eNOS), neuronal NOS and inducible NOS. On reaction with O2−, NO· forms peroxynitrite (ONOO−), a more potent form of RNS.113 Aβ peptide, which accumulates in Alzheimer’s disease (AD), generates ROS in presence of transition metal ions.114 On reaction with cellular components, accumulated ROS/RNS confer genotoxic effects and induce several DNA lesions including oxidized bases, AP sites and DNA strand breaks.115 Therefore, the cellular redox state requires balancing by the reduction of abnormally raised ROS levels. It has been demonstrated that ROS activates APE1/Ref-1 expression, and activated APE1/Ref-1 has been shown to be responsible for the adaptive response of ROS-treated cells via elevated-APE1/Ref-1-mediated increases in endonuclease activity.86 In response to oxidative stress, increases in the endonuclease activity of elevated APE1/Ref-1 have been shown to confer resistance to human glioma cells toward alkylating agents.116 It has been demonstrated that APE1/Ref-1 modulates the cellular redox state via the inhibition of intracellular ROS production or by binding with TFs, such as p53 and HIF-1α.117 A study by Hafsi and Hainaut118 reported that p53 is responsible for the regulation of redox-dependent physiological processes. Redox-sensitive p53 is regulated by ROS and in turn, activated p53 (that is, p53 that is directly activated by ROS or via redox factors, such as APE1/Ref-1) influences ROS production.119 In support of this finding, a previous study demonstrated that APE1/Ref-1 regulates cellular oxidative stress by controlling Rac1 GTPase activity and protects against oxidative cell death.120 The physical interaction between heat shock protein 70 (hsp 70) and APE1/Ref-1 was also found to protect cells against oxidative stress.121 In summary, APE1/Ref-1 regulates cellular redox states either directly or indirectly, thereby aiding the maintenance of cellular homeostasis.
Protein–protein interaction: a prerequisite for APE1/Ref-1 function
Interactions involved in DNA repair
Specific protein–protein interactions have an important role in DNA repair. Weak interactions between proteins of the BER pathway and proteins of other DNA transaction pathways can lead a decrease in BER efficiency. The repair of oxidized bases of DNA through the BER pathway depends upon interactions between APE1/Ref-1 and other downstream BER proteins. APE1/Ref-1 and pol β have been shown to interact in SN-BER using in vitro and two-hybrid studies.14 Other BER pathway enzymes with which APE1/Ref-1 interacts include PARP1, XRCC1, FEN-1 and proliferating cell nuclear antigen.12, 13, 15 PARP1 acts as a DNA damage sensor and a signaling molecule and has an important role in LP-BER, whereas XRCC1 acts as a scaffold and as a modulator that interacts with DNA repair proteins, including PARP1, ligase III and pol β in SN-BER.6, 52, 122 A study by Vidal et al.13 found that XRCC1 has an important role during the initial steps of the BER pathway by physically interacting with APE1/Ref-1 and stimulating its endonuclease and 3′-phosphodiesterase activities. In summary, coordinated protein–protein interaction(s) among BER participant proteins are beneficial because of mutual stabilization or by mutual stimulation of their enzymatic activity; in this way, the overall efficiency of the individual proteins and the entire repair machinery is improved.
Interactions involved in redox regulation and other functions
Like the repair activity of APE1/Ref-1, the redox activity of APE1/Ref-1 requires specific protein–protein interaction(s). For example, the thiol exchange reaction and the cycle involved in the reductive activation of c-Jun by APE1/Ref-1 require the specific interaction between TRX and APE1/Ref-1.68 In previous studies, NMR experiments and yeast two-hybrid systems have provided evidence for such interactions.82, 123 APE1/Ref-1 was shown to act as a reductive activator of the tumor-suppressor protein p53, which migrates to the nucleus when activated, where it activates the transcription of responsive genes.84 In an another study, APE1/Ref-1 (for redox regulation) was shown to interact stably with HIF-1α to maintain the Src-dependent, hypoxia-induced expression of vascular endothelial growth factor in pancreatic and prostrate carcinomas.124
An in vitro study showed enhanced APE1/Ref-1’s endonuclease activity at AP sites due to physical interactions between APE1/Ref-1 and hsp70 upon oxidative stress.121 In addition, APE1/Ref-1 and the WRN protein, the deficiency of which is involved in premature aging, have been shown to interact.22 APE1/Ref-1 stimulates the DNA-binding activity of p53 in a redox-independent manner by promoting the association of dimers into tetramers (that is, p53 tetramerization).125 APE1/Ref-1 has been demonstrated to interact with the p53 activator, Cdk5.28 APE1/Ref-1 has also been shown to stably interact with signal transducer activator of transcription 3 (STAT3) and p300 in some studies.47, 124, 126 In response to oxidative stress, nuclear APE1/Ref-1 interacts with ERp57, an endoplasmic reticulum (ER) protein involved in the folding and disulfide shuffling of glycoproteins and in the assembly of the major histocompatibility complex.23 Stable interaction between APE1/Ref-1 and YB-1 results in MDR-1 gene activation.25 The known protein–protein interaction(s) by which APE1/Ref-1 has a role in various biological functions are summarized in Table 1. Our recent study has identified various proteins that interact with APE1/Ref-1-interacting proteins (Figure 4) that are associated with functions required for neuronal cell survival after Aβ (25-35)-induced, stress-dependent events.26 This study shows that APE1/Ref-1 protects neurons against oxidative stress caused by Aβ in AD.

Amyloid beta (Aβ)-induced neurotoxic responses enhance the association of key neuronal proteins with apurinic/apyrimidinic endonuclease 1 (APE1)/redox effector factor-1 (Ref-1). APE1/Ref-1-interacting proteins that are differentially expressed after treatment of PC12 neuronal cells with Aβ (25–35) peptide26 include cytoskeleton elements such as tropomodulin 3 (Tmod3) and the tropomyosin alpha-3 chain; energy metabolism proteins such as pyruvate kinase M2 (PKM2); N-acetyltransferase; sulfotransferase1c; and stress-responsive proteins such as leucine-rich and death domains, anti-NGF 30 and heterogenous nuclear ribonucleoprotein-H1 (hnRNP-H1).
Biological redox balancing agents and crosstalk with APE1/Ref-1
Intracellular antioxidants
Enzymatic and non-enzymatic antioxidants provide protection against various oxidants that are generated by aerobic metabolism.127 Cellular oxidative stress results from an imbalance between oxidants and antioxidants. Antioxidants slow down or prevent oxidative reactions that generate toxic metabolites, such as free radicals, which begin a cascade of reactions that results in cell damage.128 Oxidants, such as ROS/RNS, have both beneficial and deleterious effects on organisms depending upon whether their levels are normal or abnormal.129 To maintain the appropriate balance, various non-enzymatic antioxidants (such as glutathione, vitamin C and vitamin E) and enzymatic antioxidants (such as superoxide dismutase (SOD), catalase (CAT) and peroxidases) are produced in the body. These antioxidants function in three defense processes: (1) prevention, (2) scavenging and (3) repair.127 For this, antioxidants need to interact or crosstalk with key proteins involved in these defense processes.
Crosstalk between APE1/Ref-1 and antioxidants
Limited knowledge is available regarding the direct crosstalk of APE1/Ref-1 with various cellular antioxidants. During oxidative stress in non-small cell lung cancer cells, both the downregulation of extracellular Cu-ZnSOD and CAT, and the upregulation of MnSOD together with increased APE1/Ref-1 expression levels (which are associated with responses to oxidative stress) were observed.37 APE1/Ref-1 overexpression was found to decrease further increases in intracellular superoxide levels due to tumor necrosis factor-α stimulation.130 APE1/Ref-1 overexpression did not affect the expression of Cu-ZnSOD but did decrease the expression of MnSOD significantly, indicating that APE1/Ref-1 likely has a role in regulating intracellular superoxide in the SOD-independent pathway.130 The activated tumor-suppressor gene, p53, alters the expression of antioxidant enzymes, such as MnSOD and glutathione peroxidase (GPx); however, CAT expression remains unaltered.131 In a study, it was shown that WT-p53 negatively regulates the expression of APE1/Ref-1.132 This may be the reason why p53 binds to the promoter regions for MnSOD, GPx131 and APE1/Ref-1,132 regulating their expression. In S. cerevisiae, APE1/Ref-1 regulates the transcription of some heme-binding proteins such as CAT and MnSOD.133 These studies suggest that APE1/Ref-1 is involved in regulating the antioxidant machinery of the cell.
APE1/Ref-1 and human diseases
The multifunctional enzyme APE1/Ref-1 is associated with the progression of various human diseases (Figure 5). APE1/Ref-1 can modulate susceptibility toward these human diseases via its dysregulation, post-translational modifications or polymorphism in its sequence, as observed by various research groups for several cancer types (for example, ovarian, gastro-esophageal, pancreatico-biliary, lung, prostate, cervical, colorectal, breast, hepatocellular, bladder, head and neck, gastric, and glial cancers).71, 134 The same is true for diseases such as CVD,31, 71, 135 AD,26, 27, 28, 136 Parkinson’s disease (PD),28, 137 Huntington’s disease (HD),138 amyotropic lateral sclerosis (ALS),139 cerebral ischemia140 and HIV pathogenesis.141 These studies underscore the diverse functional association of APE1/Ref-1 with various human diseases, indicating that this is a promising therapeutic research area for the treatment and management of human diseases.

Apurinic/apyrimidinic endonuclease 1 (APE1)/redox effector factor-1 (Ref-1) has a pivotal role in various human diseases. Owing to its vital role in the BER pathway, in balancing intra-cellular redox states and in the reductive activation of various transcription factors (TFs) controlling cell survival pathways, alterations in APE1/Ref-1 expression and functions have a crucial role in various human diseases including cancer, cardiovascular disease (CVD) and neurodegenerative diseases (NDs). The overexpression and enhanced enzymatic activities of APE1/Ref-1 have been linked with the conferral of survival advantage and chemoresistance in cancer cells. APE1/Ref-1 also has an important role in CVD by governing H-Ras-mediated endothelial nitric oxide synthase (eNOS) activity and NO bioavailability. In NDs, such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and amyotropic lateral sclerosis (ALS), decreased neuronal expression of APE1/Ref-1 after neuronal insult decreases cell viability and promotes neurodegeneration. In addition to alterations in the expression of APE1/Ref-1 and its subcellular localization and enzymatic functions, single-nucleotide polymorphisms (SNPs) in the APE1/Ref-1 gene have been also reported to have a role in ALS, AD and cancer. In addition, various post-translational modifications (PTMs), such as acetylation,7 ubiquitination,93, 100 s-nitrosylation,234 sumoylation21 and phosphorylation,235 have been demonstrated to regulate APE1/Ref-1 functions in various human diseases.
Perspectives on NDs
Alzheimer’s disease
Defective BER pathway proteins and functions have been linked to age-associated neurological disorders.5 AD, the most common form of age-related dementia, is a progressive and fatal disorder that is clinically characterized by memory loss and behavioral abnormality. Pathologically, AD can be recognized by the deposition of Aβ in neuronal cells.28 In the AD brain, APE1/Ref-1 expression increases in the region of neuronal injury. The p35 activator Cdk5 interacts directly with APE1/Ref-1, resulting in the phosphorylation of Thr232 of APE1/Ref-1.28 When Thr232 is phosphorylated, APE1/Ref-1 exhibits decreased endonuclease activity (compared with the non-phosphorylated form), which is associated with neuronal death in PD116 and AD.28 It has been shown that persistent exposure to 5 μM Aβ (1–42) for >72 h is associated with a decrease in APE1/Ref-1 expression.30 APE1/Ref-1 expression was found to be increased in the nuclear extracts of AD patients compared with age-matched controls.27 Another immunohistochemical study showed high levels of nuclear APE1/Ref-1 expression in the cerebral cortex of AD patients, supporting a role for APE1/Ref-1 in cellular adaptation to oxidative stress.136 Recently, APE1/Ref-1 has been found to be upregulated after glutamate-induced oxidative DNA damage, and Ca2+-induced, cAMP response element-binding (CREB)-mediated APE1/Ref-1 expression has been suggested to be important for neuronal survival.142 A recent review by Mantha et al.5 discussed the association of single-nucleotide polymorphisms (SNPs) of BER enzymes (including APE1/Ref-1) with the development of AD.
Parkinson’s disease
PD is a common motor neurodegenerative disorder that is associated with a progressive loss of dopaminergic (DRG) neurons in the substantia nigra and locus coeruleus. Multiple genetic and environmental factors are correlated with the pathology of PD.137 Genetic variants of APE1/Ref-1, XRCC1 and XRCC3 are possible risk factors for increased oxidative stress that may cause a loss of DRG neurons in the substantia nigra and locus coeruleus, leading to abnormal signal transduction and ultimately, to the development of PD.137 Phospho-APE1/Ref-1 immunoreactivity has been observed in the nuclei of DRG neurons from post-mortem tissues of patients with PD, and nuclear extracts were shown to have decreased endonuclease activity due to post-translational modification (phosphorylation).28
Huntington’s disease
HD is a neurodegenerative disorder that is clinically characterized by involuntary choreiform movements, behavioral abnormalities and dementia. Pathologically, this disorder is characterized by neuronal loss in the striatum and cerebral cortex.143 Given the importance of APE1/Ref-1 for the maintenance of mitochondrial function, decreased APE1/Ref-1 repair activity during mitochondrial DNA damage repair may have a role in the mitochondrial dysfunction associated with HD.138 In addition, increased nuclear and mitochondrial localization of APE1/Ref-1 after exogenous oxidative stress has been observed in neuronal cells. Cells expressing a mutant form of huntingtin exhibit higher lesion occurrence and decreased lesion repair, or both; intriguingly, silencing of APE1/Ref-1 in HD cells (but not in WT cells) was found to exacerbate mitochondrial dysfunction, suggesting that APE1/Ref-1 has a critical role in maintaining mitochondrial function.138
Amyotrophic lateral sclerosis
ALS is another ND that is characterized by progressive weakness and muscular atrophy. An elevated concentration of intracellular-free radicals that leads to motor neuron degradation has been identified as one of the causal factors of this disease.144 In sporadic ALS, reduced expression and repair activity of APE1/Ref-1 have been observed.145 In an another study, APE1/Ref-1 levels were found to be elevated in the brain and spinal cord samples of patients with ALS, indicating the possible activation of DNA damage repair mechanism(s) through the BER pathway; this activation may be an upstream mechanism for motor neuron degeneration in ALS139 in cases where the mechanisms for DNA repair may be exhausted upon the prolonged stimulation of degenerative cascades. In addition, analyses of cloned APE1/Ref-1 transcripts from nine ALS patients and two patients (twins) with familial ALS revealed five-amino-acid substitutions: Leu104Arg, Glu126Asp, Asp148Glu, Asp283Gly and Gly306Ala.146 All of these SNPs were observed once, excluding Glu126Asp, which was reported in both twins with familial ALS; however, no amino-acid changes in APE1/Ref-1 were detected in five healthy controls. In an independent study, an APE1/Ref-1 Asp148Glu variant, which was found in both ALS and control samples, was shown to be associated with sporadic but not familial ALS.146, 147
Cerebral ischemia
Cerebral ischemia is caused by a blood supply to the brain, which results in oxidative stress in neuronal cells. In a study, elevated levels of APE1/Ref-1 were correlated with neuronal survival against ischemic insult, although the results depended on the central nervous system (CNS) region examined, the protective treatment applied and the degree of insult.140 Neurons with decreased APE1/Ref-1 expression and endonuclease activity were found to be extremely vulnerable to cell death induced by in vitro ischemia, indicating that oxidative base lesions and AP sites can trigger ischemic cell death.148 In another study, oxidative stress-mediated increased APE1/Ref-1 expression was observed in the hippocampus of rat brain in response to global cerebral ischemia/traumatic brain injury/cold injury; this observation was associated with neuronal apoptosis in rats.149, 150
Perspectives on cancer
Cancer is defined as the uncontrolled growth of abnormal cells in the body and is caused by abnormalities in various mechanisms including deletion, amplification or mutation in the genetic material. Two principal modalities for the prevention and cure of cancer exist; early detection and efficient treatment. First, for early detection, possible candidate genetic markers should exhibit altered expression patterns in cancerous cells when compared with healthy normal cells. Second, for prevention and treatment, two further main approaches are used to prevent or cure cancer: (a) obstructing the cancer at the very first error that occurs during the initial phase (that is, at initiation) and (b) forestalling all the possible molecular pathways by which the cancer sustains its own survival (that is, during growth, progression and metastasis).151 The first approach, which appears more direct and obvious than the later, and perhaps is more difficult to achieve, hence not yet been developed for therapeutic use. Therefore, the second approach is exploited more widely in current research. Moreover, blocking a single molecular pathway is insufficient in most cases because different types of cancer appear to be heterogeneous. Therefore, targeting multiple molecular pathways underlying cancer survival can result in improved therapeutic intervention. APE1/Ref-1 can be used as both a genetic marker and a molecular target. However, a problem arises when WT APE1/Ref-1 enzyme is overexpressed in cancer cells exhibits altered repair and redox functions under such conditions. Thus, APE1/Ref-1 helps cancer cells to survive; thus, it confers resistance to them against chemotherapy/radiotherapy.152
The expression pattern and subcellular localization of APE1/Ref-1 in relation to cancer
Dysregulation of APE1/Ref-1 has been reported to be associated with cancer. Evidence from immunohistochemical analyses has demonstrated that alterations in both subcellular distribution and expression levels are linked with numerous cancers.32, 33, 34, 35, 36, 37, 153 A positive correlation was also found between the endonuclease activity of APE1/Ref-1 and tumor grade.154 Various studies using APE1/Ref-1-small interfering RNA (siRNA) in cancer cell lines overexpressing APE1/Ref-1 have demonstrated that APE1/Ref-1 has a role in cancer development and progression. One study has found a significant association between high levels of APE1/Ref-1 expression and osteosarcoma risk. This study also showed increased chemosensitivity in an osteosarcoma cell line (human osteogenic sarcoma) after the use of APE1/Ref-1-siRNA.155
The coordination between the expression pattern of APE1/Ref-1 and its subcellular location has differed among different cell types studied thus far. Normally, APE1/Ref-1 is predominantly expressed within the nucleus rather than in the cytoplasm. Cytoplasmic expression is mainly observed in cell types exhibiting active metabolism associated with the cell organelles mitochondria and ER, in response to oxidative stress.88 Altered APE1/Ref-1 subcellular localization patterns (mixed localization or predominant nuclear or cytoplasmic localization) have been observed in many cancers. Positive nuclear APE1/Ref-1 expression has been observed in head and neck cancer, in rhabdomyosarcomas, bladder, ovarian, gastro-esophageal and pancreatico-biliary cancers.32, 156, 157, 158 Positive cytoplasmic APE1/Ref-1 expression has been observed in some cancers, such as thyroid, prostate and hepatocellular cancers.33, 36, 159 More localized distribution of APE1/Ref-1 was observed in non-small cell lung cancer cell.37 In addition, upregulated APE1/Ref-1 expression levels were found to be associated with poor prognosis in medulloblastoma.160 A positive correlation between aggressive tumor grade and nuclear APE1/Ref-1 expression in ovarian, gastro-esophageal and pancreatico-biliary cancers was observed.32 Nuclear localization was also found to be associated with cellular differentiation pattern and lymph node status.36 In pancreatic cancer cells, APE1/Ref-1 also regulates the DNA-binding affinity of TFs, such as STAT3, without affecting the other mechanisms by which it is regulated (that is, phosphorylation, nuclear translocation and protein level). The APE1/Ref-1 redox inhibitor E3330 also inhibits the transcriptional activity of STAT3.161 E3330 inhibits the proliferation of pancreatic cancer cells by arresting the cell cycle without inducing apoptosis and decreases the transcriptional activity of NF-κB, HIF1α and AP-1, which are regulated by APE1/Ref-1.162 Taken together, the results of these studies suggest that alterations in APE1/Ref-1 expression levels and subcellular distribution can be used as a significant prognostic biomarker for various cancers.
The efficacy of DNA-damaging agents in cancer treatment is diminished by the increased repair and redox activities of APE1/Ref-1 in cancer cells. Elevated APE1/Ref-1 levels are correlated with chemoresistance and radioresistance; therefore, recent investigations have focused on identifying APE1/Ref-1 as a potential promising target for sensitizing cancer cells to chemotherapy and radiotherapy.163, 164 These findings are also suggestive that APE1/Ref-1 expression levels can be used as a predictive marker for treatment response.163 Although immunohistochemistry analyses and siRNA knock-down studies are able to provide sufficient information regarding the role of APE1/Ref-1 in normal versus cancerous cells, these studies do not reveal the significance of the individual repair or redox activities of APE1/Ref-1. However, the identification of causal activity (modulated repair or redox activity) in cancer risk is critical for further research and therapeutic applications.
Malignant gliomas are most common primary, intracranial tumors in adults and are often fatal forms of brain cancer, with a 2-year survival rate of ∼26%.165 Treatment options for gliomas include surgical resection followed by chemotherapy or radiation, both of which generate lesions that are repaired by different repair pathways. The high-throughput screening of 60 000 small-molecule compounds has led to the identification of several small-molecule inhibitors of endonuclease activity. Of these screened small compounds, ARO3 stands out the most promising inhibitor of endonuclease activity in glioma therapy.165 Substantially elevated levels of APE1/Ref-1 and enhanced endonuclease activity are characteristic of adult gliomas.154 APE1/Ref-1 endonuclease activity was found to be significantly greater in high-grade tumors than in low-grade tumors.154 Glial cell-derived neurotrophic factor (GDNF) is a neurotropic factor that is specific for the survival and differentiation of mid-brain DRG neurons. It has been reported that APE1/Ref-1-stimulated, GDNF/GDNF family receptor alpha 1 (GFRα1)-mediated downstream signaling enhances the proliferation of neuroblastoma cells;166 challenging the current therapeutic strategies used against gliomas.
To identify the role of APE1/Ref-1 repair activity in cancer progression, certain selective inhibitors have been used. In one such study, APE1/Ref-1’s repair activity was found to provide resistance to glioblastoma cells against alkylating agents.116 In another study, it was found that the inhibition of endonuclease activity sensitized breast cancer cell lines to temozolomide.152 The novel small-molecule-based inhibitor of APE1/Ref-1’s redox activity, E3330, was found to block the redox activation of the TF AP-1; thus, E3330 increased the sensitivity of ovarian cancer cells toward alkylating agents.167 It was further demonstrated that when administered along with methoxyamine, which blocks repair activity, tumor cell killing is markedly enhanced.167 It was also found that E3330 abolishes the role played by APE1/Ref-1 in angiogenesis.76 Further research utilizing selective inhibitors against the individual functions of APE1/Ref-1 is required to delineate the underlying cause to enhance the current therapeutic potential toward various cancers.
APE1/Ref-1 polymorphism and cancer susceptibility
APE1/Ref-1 polymorphism can increase cancer risk. The probable mechanism underlying the association of APE1 gene polymorphism and cancer risk is that these SNPs lead to amino-acid substitutions, which may result in alterations of the functions of APE1/Ref-1.146 Until now, some APE1/Ref-1 variants have been identified as likely to have a role in cancer development and progression. Asp148Glu (T/G, codon 148, exon 5, Asp to Glu) is a commonly found variant of APE1/Ref-1; this mutation is located in the DNA-repair region of the APE1/Ref-1 gene. This mutation is found among different populations with high frequency and has been associated with various tumors and NDs.168, 169, 170, 171, 172, 173 Another SNP, 656 T>G, which lies in the promoter region of the APE1/Ref-1 gene, is associated with lung cancer risk.174 The homozygous SNP, 656 T>G, is associated with renal cell carcinoma.175 Reduced endonuclease activity has been reported in four of seven identified APE1/Ref-1 variants.146 Most of these studies were population based, so it is possible that certain factors such as environmental conditions, dietary lifestyle or heterogeneity among populations can act as effect modifiers.146 Therefore, further studies examining the functional and biochemical aspects of APE1/Ref-1 variants that are associated with cancer development and progression are warranted. Such studies may unveil the potential role of APE1/Ref-1 polymorphism in predisposing toward various cancers and aid in prognosis and prediction. Thus, APE1/Ref-1 has the potential to act as a predictive, prognostic and therapeutic target for various cancers.
Perspectives on CVDs
CVDs are the leading cause of death worldwide; the major manifestations of CVD include myocardial infarction, coronary artery disease, stroke and peripheral artery disease.176 During 2008, 244 deaths were recorded because of CVD per 10 000 individuals according to a report published by the American Heart Association.177 Hypertension affects >50 million Americans and 65% of people older than 65 years.178 Various DNA-repair gene polymorphisms are associated with CVD.179 Several studies have shown an association between the DNA-repair enzyme APE1/Ref-1 and CVD, but the molecular mechanism(s) involved require further study.
In an investigation performed on aortic coarctation-induced hypersensitive rat models, APE1/Ref-1 expression was found to be elevated.31 In an another study, an association between APE1/Ref-1 gene polymorphism and essential hypertension was observed.135 A study was carried out using the COS-7 cell line and an APE1/Ref-1+/− mouse model to study the role of endogenous APE1/Ref-1 in the regulation of endothelium-dependent vascular tone and systemic blood pressure.71 Damaged endothelium-dependent vascular relaxation due to reduced eNOS activity and basal endothelial NO production was observed in APE1/Ref-1+/− mice relative to WT mice.71 The stimulation of eNOS activity was not caused by APE1/Ref-1 overexpression. However, the nuclear localization of APE1/Ref-1 was critical for the stimulation of eNOS expression. Finally, the mechanism by which APE1/Ref-1 governs endothelium-dependent vascular tone and systemic blood pressure was studied. Decreased APE1/Ref-1 redox activity with respect to the H-ras-mediated, PI3 kinase/AKT pathway-dependent Ca2+ sensitization of eNOS was suggested as a possible cause of decreased basal endothelial NO production and subsequent damaged-endothelium-dependent vascular relaxation.71 APE1/Ref-1 was also reported to cause Ca2+-mediated repression of the renin gene, which is a crucial part of the renin–angiotensin system and a major regulator of blood pressure. The results further indicated that the Ca2+-mediated association of APE1/Ref-1 with the corepressor histone deacetylase 1 complex led to recruitment at the rennin-enhancer region and thus had a role in regulating blood pressure.180
Perspective on other diseases
APE1/Ref-1 has been shown to have functions in diseases other than neurodegenerative disorders, cancer and CVD, including bacterial Helicobacter pylori (H. pylori) infection. APE1/Ref-1 expression was found to be elevated during H. pylori infection in human gastric epithelial cells (GECs). Using siRNA technology, it was demonstrated that the silencing of APE1/Ref-1 in human GECs decreased the H. pylori-mediated activation of AP-1 and NF-κB. Consequently, silencing of APE1/Ref-1 resulted in decreases in interleukin-8 mRNA and protein expression. However, in contrast, overexpression of APE1/Ref-1 reversed the above phenotype.181 One study has demonstrated the role of APE1/Ref-1 acetylation in regulating H. pylori-induced apoptosis in GECs.182 A dual role of APE1/Ref-1 (in terms of DNA repair and acetylation-mediated functions) in the modulation of the intrinsic and extrinsic apoptotic pathways was identified earlier.183 However, mutant versions of APE1/Ref-1 (the DNA repair variant His309Asn and the non-acetylable variant Lys6Arg/Lys7Arg) were unable to decrease apoptosis. Together, these findings suggested that the DNA-repair function of APE1/Ref-1 has a role in regulating the mitochondrial caspase-9-mediated apoptotic pathway, whereas the acetylation function of APE1/Ref-1 regulates the caspase-8-dependent extrinsic apoptotic pathway.183
APE1/Ref-1 is also associated with HIV pathogenesis. Certain cytosolic factors are present in host cells that help to prevent the autointegration of HIV DNA, a type of suicidal pathway for HIV that can halt infection. One such cytosolic factor is APE1/Ref-1, which acts as a part of the ER-associated DNA-repair ‘SET complex’ along with other nucleases (NM23-H1) and 3′-repair exonuclease 1. This complex binds to HIV DNA in the cytosol, preventing viral autointegration and facilitating infection. Thus, the APE1/Ref-1-containing ‘SET complex’ increases the efficiency of successful viral integration with the host chromosome.141 Further studies are required to understand the exact role played by APE1/Ref-1 through its repair and redox functions in HIV pathogenesis.
APE1/Ref-1 can also act as a promising candidate in preventing the neurotoxicity caused by cancer therapeutics in terms of the attenuation or reversal of therapy-induced damage.38 Englander39 reported the overexpression and localization of DNA repair proteins in DRG neurons damaged by antimitotic agents. These authors suggested that such DNA-repair proteins can be used to predict post-treatment risks in PNS neurons and can help to overcome neurological side-effects such as CIPN that has been caused by cancer therapeutics. Further studies are required to determine new therapeutic strategies that may aid in enhancing the DNA repair functions of nuclear and mitochondrial BER enzymes in neurons.5
Another role of APE1/Ref-1 has been observed in age-related macular degeneration and other eye-related diseases. Neovascularization in the retina is a key pathology in several ocular diseases, such as age-related macular degeneration, diabetic retinopathy and retinopathy of prematurity,184 and is the main cause of severe vision loss. Studies have shown that APE1/Ref-1 is highly expressed in the retina and choroid and in retinal vascular endothelial cells in mice; these studies have provided evidence that APE1/Ref-1 redox activity is required for efficient retinal endothelial cell proliferation, migration and tube formation.184, 185 Using small-molecule inhibitors, APE1/Ref-1 ’s redox activity was found to cause retinal angiogenesis and is thus a possible therapeutic target in neovascular age-related macular degeneration and other eye-related diseases. The specific redox activity inhibitor E3330 (recently renamed APX3330) has been used to elucidate the role of APE1/Ref-1’s redox function, both in vitro (in retinal vascular endothelial cells) and in vivo (in a very-low-density-lipoprotein-(VLDL)-receptor knockout mouse (Vldlr−/−) model). Retinal vascular endothelial cells exhibited inhibited proliferation, migration and tube formation. Vldlr−/− mice exhibited decreased angiomatous proliferation (that is, retinal angiomatous proliferation (RAP)-like neovascularization).184 The investigation of APE1/Ref-1 can be further widened to other areas.
Importance of phytochemicals in modulating the functions of APE1/Ref-1 toward the goal of developing therapeutic interventions for human diseases
Dietary substances have an important role in improving the health status of patients suffering from various diseases. Supplementation of the diet with various phytochemicals has been shown to enhance the response of routinely used drugs in treatment strategies. Phytochemicals have been shown to hold the potential to alter the repair and redox activities of APE1/Ref-1.186 The modulation of the expression, repair and regulatory functions of APE1/Ref-1 by plant components for the treatment and prevention of various human diseases is reviewed in this section (Figure 1).
Soy isoflavones
Radiation therapy is used for the effective treatment of cancer but has adverse effects on normal cells. Soy isoflavones have been reported to exert a cytotoxic effect on cancer cells187 and can be used as dietary modulators for the treatment of cancer and other diseases. Increased DNA-repair enzyme activity has been observed in lung cancer cells, and complementary treatment with soy isoflavones rendered these cells more sensitive to radiation therapy.186 Inhibition of the radiation-enhanced repair and redox activities of APE1/Ref-1 promoted the effective use of isoflavones in association with radiotherapy.188 Simultaneous treatment with soy isoflavones not only decreases the survival rate of cancer cells but also prevents other cells from the adverse effects of radiotherapy by improving their physiological state.189 This may be due to increased APE1/Ref-1 repair activity in the normal cells (which contrasts with its downregulation in cancer cells). However, no study has measured the APE1/Ref-1 levels thus far. Genistein and daidzein are components of soy extracts that exhibit contrasting properties. Genistein increases metastasis whereas daidzein inhibits metastasis in the lymph nodes by affecting APE1/Ref-1 levels and the levels of other TFs in prostate cancer cells.190 In contrast, genistein when used alone, sensitizes prostate cancer cells to radiotherapy.191 Therefore, pure components and their analogs could be used as drug targets in the development of cancer therapeutics. In addition, APE1/Ref-1 downregulation by soy extracts rendered pancreatic and oral cancer cells more responsive toward radiotherapy.134, 187
Further experimental studies are advocating for the role of soy isoflavones in the improvement of memory and mental health.192 Soy isoflavones can prevent oxidative stress and reduce the damaging effects caused by aging and AD.193 Dose-dependent effect of soy isoflavones was observed in D-galactose-treated, aging-induced C57BL/6J (B6) mice, where DG-induced Aβ and presenilin I expression was found lowered by soy isoflavones in the B6 mice.193 Genistein also showed protective effect on the cortical neurons with iron-induced lipid peroxidation and toxicity.194 These soy isoflavones proved to provide neuroprotection under both in vitro and in vivo conditions by ER-mediated pathway and tyrosine kinase inhibition.195 Although the role of APE1/Ref-1 has not been studied in this regard, it may be possible that APE1/Ref-1’s functions are engaged in neuroprotection.
Resveratrol
Resveratrol (trans-3′,4′,5-trihydroxystilbene) is an important polyphenolic antioxidant present in grapes and other dietary components.153 Resveratrol exhibits antiproliferative activity against cancer cells, which has been well elucidated using in vitro studies.153, 196, 197 Melanoma cells express APE1/Ref-1 at greater than basal levels. Cells treated with resveratrol exhibit reduced AP-1 and NF-κB DNA-binding activity. It has been reported that the AP-1 DNA-binding activity of APE1/Ref-1 was significantly reduced in a dose-dependent manner by direct co-incubation with resveratrol. Upon APE1/Ref-1 depletion by immunoprecipitation, no further inhibition was evident in the resveratrol treatment. Therefore, the inhibitory effects of resveratrol on APE1/Ref-1 occurred mainly due to its redox function.153 In addition, it was also found that the dose-dependent inhibition of APE/Ref-1 expression by resveratrol rendered melanoma cells are more susceptible to the action of drugs such as dacarbazine.153 Resveratrol also has an important role in the prevention of stress by modulating APE/Ref-1.198 Aluminum chloride (AlCl3)-induced neuroinflammation in rat brain was prevented by treatment with resveratrol, and elevated levels of APE1/Ref-1 were suggested as the main cause of the prevention of toxicity to the neuronal cells, together with the expression of tumor necrosis factor-α, interleukin-6 and inducible NOS.198
Furthermore, resveratrol was also found to have preventive and therapeutic value for the treatment of CVD.199 It has been established that resveratrol has a role in the prevention of myocardial infarction by maintaining the redox environment during stem cell regeneration in rats.200 In the presence of resveratrol, induced myocardial ischemia in rats resulted in the regeneration of the myocardium by the stem cells, which would not survive for long in the absence of resveratrol. The resveratrol-induced utilization of APE1/Ref-1 proved responsible/beneficial for maintaining the redox environment in the cells.200 Supplementation of resveratrol in patients suffering from CVD enhanced the effectiveness of stem cell therapy.199 Resveratrol also prevented hypertrophy in rats201 by activating NOS activity, indicating that APE/Ref-1 has a regulatory role in such prevention. Furthermore, rats that were treated with resveratrol exhibited enhanced DNA repair activity because of the upregulated expression of APE1/Ref-1, thus overcoming myocardial stress.202
Curcumin
Curcumin is the active component of Curcuma longa, which is widely used as a spice and in Indian Ayurvedic medicines. This substance acts as a potent anti-inflammatory agent because of its antioxidant activity and ROS scavenging properties.203 Curcumin’s key property of preventing oxidative damage has been implicated in controlling and preventing various human diseases.204, 205, 206 Curcumin acts by inducing apoptosis or cell cycle arrest in cancer cells207 and has undergone phase 1 clinical trials for cancer prevention; in these trials, curcumin was found to be safe at intakes of up to 8 g per day.208 One of the mechanisms through which curcumin displays its anticancer properties is through the inhibition of the mitogen activated protein kinase kinase kinase-Jun-NH2 kinase (MEKK1-JNK) pathway by suppressing the activity of AP-1 and NF-κB signaling molecules.209 Curcumin may block the downstream signaling cascades of AP-1 and NF-kB by inhibiting the redox function of APE1/Ref-1. The main disadvantage of curcumin use is that the molecule is rapidly metabolized, thereby decreasing its bioavailability. To overcome this problem, an alternative approach has been suggested to increase the bioavailability of curcumin inside the body by supplementing it with piperine.210 The elevated expression of APE1/Ref-1 in cancer cells leads to the development of drug resistance. Curcumin decreases APE1/Ref-1 levels in cancer patients.211 The combined treatment of epigallocatechin-3 gallate (EGCG, a constituent of tea) and curcumin in follicular lymphoma patients decreased the levels of APE1/Ref-1 and TFs to much larger extent than other drugs routinely used in the treatment of this disease.211 In addition, curcumin exhibits a hepato-protective role and prevents CCl4-induced fibrosis in rat models.211 Therefore, it can be inferred that combination therapy is beneficial and that dietary supplementation is widely important in cancer therapy.
The accumulation of ROS in response to metals results in the oxidation of nitrogenous bases that are present in DNA. Metals such as copper (Cu) and iron (Fe) interact with DNA repair enzymes and oxidize DNA bases; thus, these metals are implicated in NDs.212 Curcumin acts as a strong metal chelator and has the ability to reverse the inhibition of the NEIL1 caused by metals, such as Cu and Fe, which adversely affect DNA repair in vitro.212 Curcumin exhibits dual advantages as a potential therapeutic agent by reducing oxidative stress and acting as a metal chelator. Oxidative damage is a common cause of AD, which is associated with the deposition of Aβ; as an antioxidant, curcumin has the potential to reduce such stress in AD.211 In low doses, curcumin reduces the formation of Aβ (in both soluble and insoluble forms) and plaque formation in transgenic AD mice.213 Moreover, curcumin has a strong ability to cross the blood–brain barrier, enabling it to decrease the aggregation of fibrils, resulting in the disaggregation of the Aβ and rendering it effective for the prevention and treatment of AD.214
(−)–Epigallocatechin-3 gallate
Catechins isolated from the leaves of green tea (Camellia sinensis) have a number of beneficial health effects, including anticarcinogenicity, as demonstrated in various tumor models.215 EGCG has been shown to block the interaction between mouse double minute 2 homolog (MDM2) and p53, thus inhibiting MDM2-mediated p53 ubiquitination, which directly affects DNA damage repair.216 EGCG exhibits significantly greater anti-invasive properties than other catechins. EGCG reverses the epithelial-to-mesenchymal transition in melanoma cells.217 EGCG, when used in combination with curcumin, decreased the levels of APE1/Ref-1 and induced complete remission in B-cell non-Hodgkin’s lymphoma patients.215 EGCG was reported to interface with NF-κB activation, thereby resulting in an increased tumor cell response to various NF-κB-activating anticancer drugs, including doxorubicin. Furthermore, EGCG has been shown to inhibit cell migration in melanoma cells after 24 h of treatment in a dose-dependent manner and has been proposed to be associated with cycloxygenase-2 expression and prostaglandin E2 production.217
Decursin
The root of Angelica gigans Nakai (Umbelliferacea) is used in traditional oriental herbal medicine to treat female afflictions and is regarded as a version of ginseng appropriate for females. The pyranocoumarin decursin is one of the main active constituents of Angelica gigans Nakai.218 Decursin is able to cross the blood–brain barrier and penetrates the CNS. Decursin exhibits neuroprotection against Aβ-induced oxidative stress in PC12 cells via nuclear factor-like 2/NF-E2-related factor 2-associated antioxidant-responsive elements.219 Nuclear factor 2/NF-E2-related factor 2 has been shown to regulate and control the expression of many detoxifying genes, including SOD, CAT, GPx and glutathione S-transferase (GST).219 Treatment with decursin (0.01–10 μM) has been shown to protect PC12 cells against Aβ-induced neurotoxicity.220 Further, it has been demonstrated that Decursin inhibits the growth and proliferation of human umbilical vein endothelial cells in a dose-dependent manner by delaying the exit from the G1 phase, thus suppressing angiogenesis.221 Studies are needed to test the effect of decursin on Aβ-induced neurotoxicity and its likely role in modulating the functions of APE1/Ref-1 in experimental AD model systems as a potential therapeutic agent because of its ability to cross the blood–brain barrier.222
Polyphenols (quercetin, luteolin and rosmarinic acid)
Quercetin, luteolin and rosmarinic acid are polyphenolic phytochemicals that are present in a variety of food products, including onions, citrus fruit, berries, buckwheat, apples, tea and wine.223 The protective effect against oxidative DNA damage acts either by preventing the oxidation of DNA (acting as an antioxidant) or by enhancing the frequency of repair by stimulating repair enzymes. The antioxidant properties of quercetin and rosmarinic acid have been examined in various studies.224, 225, 226 Quercetin protects DNA in Caco-2 cell lines against oxidative DNA damage.227 Similar results have been found using polyphenols in murine leukemia L1210 cells, HMB2 cells and HepG2 cells.228, 229, 230 Rosmarinic acid is also capable of protecting against doxorubicin-induced DNA damage in V79 cells.231 When tested in PC12 cells, the polyphenolic compounds quercetin, luteolin, and in particular, rosmarinic acid conferred protection against oxidative-stress-induced DNA damage by increasing the expression levels of DG 8-oxoguanine glycosylase, thus increasing repair capacity.232 In contrast, it was found that treatment with rosmarinic acid alone decreased the expression of APE1/Ref-1.232 In addition, rosmarinic acid and luteolin protected DNA against oxidative damage in Caco-2 cells and in HeLa cells by increasing DNA repair capacity.233 Together, these studies show that polyphenols can enhance DNA repair activity and prevent oxidative DNA damage, thereby protecting against oxidative DNA damage by modulating the activities of APE1/Ref-1. Further biochemical and molecular studies are required to understand the mechanism of action of these polyphenols and their effect on modulating the activities of APE1/Ref-1.
Conclusions and future perspectives
APE1/Ref-1 was primarily discovered as an enzyme that repairs oxidative DNA damage through its participation in the BER pathway. Later, its other major functions in the redox regulation (Ref-1 activity) of various TFs involved in cell cycle control, drug resistance, angiogenesis, inflammation, cell survival pathways, cellular homeostasis regulation, acetylation-mediated (redox-independent) transcriptional regulation and interactions with specific proteins involved in diverse trans-acting cellular pathways were also discovered. The results from various studies have confirmed that APE1/Ref-1’s pleiotropic functions are crucial for promoting cell survival and maintaining cellular genomic integrity. Evidence from various molecular studies suggests that differences in the expression pattern and subcellular localization of APE1/Ref-1 are associated with various human pathologies, including neurodegenerative disorders, cancer, CVD and other diseases, such as H. pylori and HIV infections. Studies demonstrating the exploitation of APE1/Ref-1 as a therapeutic target for cancer treatment have used approaches such as APE1/Ref-1 siRNA silencing technology or APE1/Ref-1 overexpression in cancer cell lines. These techniques do not affect the repair and redox functions of APE1/Ref-1 individually; thus, it is difficult to interpret the target functions with a view to developing appropriate therapeutic agents. Studies exploiting small molecules that inhibit the repair or the redox activity of this protein separately are emerging as promising tools. However, new approaches are required to elucidate (a) the underlying mechanisms by which APE1/Ref-1 confers chemo- and radio resistance to tumor cells and (b) the signaling mechanism(s) that regulate the expression pattern and subcellular localization of APE1/Ref-1. Some epidemiological studies have linked APE1/Ref-1 gene polymorphism with various cancers. The presence of effect modifiers, such as heterogeneity among populations, environmental exposure and dietary lifestyle and the lack of sufficient functionally relevant biochemical evidence have limited the association of APE1/Ref-1 polymorphism with particular cancers. Therefore, further in vitro biochemical studies are required to clarify possible relationships. Proteomic studies can help to determine the specific APE1/Ref-1-mediated redox-dependent and redox-independent protein–protein interactions that occur in response to exogenous and endogenous oxidative stress and would aid in the development of effective therapeutic strategies. The use of various plant products to treat human diseases is an ancient practice. Natural plant products and phytochemicals, such as soy isoflavones, curcumin, resveratrol, tea polyphenols and decursin can modulate the repair or redox activities of APE1/Ref-1 or both, as well as the interaction between APE1/Ref-1 and other proteins. The use of these phytochemicals as adjuvants in conjunction with standard therapies and as antioxidants for use in normal healthy cells is a promising approach for the treatment and prevention of diseases. Further in vitro studies analyzing the differential molecular effects of phytochemicals on the expression pattern, individual repair and redox activities and protein–protein interactions of APE1/Ref-1, as well as subsequent in vivo investigations are needed to develop future applications of these phytochemicals as APE1/Ref-1-mediated interventions for various human diseases.
References
Barnham KJ, Masters CL, Bush AI . Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 2004; 3: 205–214.
Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA . Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009; 30: 2–10.
Cooke MS, Evans MD, Dizdaroglu M, Lunec J . Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 2003; 17: 1195–1214.
D'Errico M, Parlanti E, Dogliotti E . Mechanism of oxidative DNA damage repair and relevance to human pathology. Mutat Res 2008; 659: 4–14.
Mantha AK, Sarkar B, Tell G . A short review on the implications of base excision repair pathway for neurons: relevance to neurodegenerative diseases. Mitochondrion 2013; 16: 38–49.
Hegde ML, Hazra TK, Mitra S . Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res 2008; 18: 27–47.
Bhakat KK, Mantha AK, Mitra S . Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid Redox Signal 2009; 11: 621–637.
Barzilay G, Hickson ID . Structure and function of apurinic/apyrimidinic endonucleases. Bioessays 1995; 17: 713–719.
Xanthoudakis S, Miao GG, Curran T . The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc Natl Acad Sci USA 1994; 91: 23–27.
Chou KM, Cheng YC . An exonucleolytic activity of human apurinic/apyrimidinic endonuclease on 3′ mispaired DNA. Nature 2002; 415: 655–659.
Hegde ML, Izumi T, Mitra S . Oxidized base damage and single-strand break repair in mammalian genomes: role of disordered regions and posttranslational modifications in early enzymes. Prog Mol Biol Transl Sci 2012; 110: 123–153.
Khodyreva SN, Prasad R, Ilina ES, Sukhanova MV, Kutuzov MM, Liu Y et al. Apurinic/apyrimidinic (AP) site recognition by the 5′-dRP/AP lyase in poly (ADP-ribose) polymerase-1 (PARP-1). Proc Natl Acad Sci USA 2010; 107: 22090–22095.
Vidal AE, Boiteux S, Hickson ID, Radicella JP . XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein–protein interactions. EMBO J 2001; 20: 6530–6539.
Bennett RAO, Wilson DM, Wong D, Demple B . Interaction of human apurinic endonuclease and DNA polymerase β in the base excision repair pathway. Proc Natl Acad Sci USA 1997; 94: 7166–7169.
Dianova II, Bohr VA, Dianov GL . Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry 2001; 40: 12639–12644.
Bapat A, Fishel ML, Kelley MR . Going Ape as an approach to cancer therapeutics. Antioxid Redox Signal 2009; 11: 651–667.
Ema M, Hirota K, Mimura J, Abe H, Yodoi J, Sogawa K et al. Molecular mechanisms of transcription activation by HLF and HIF1α in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J 1999; 18: 1905–1914.
Gaiddon C, Moorthy NC, Prives C . Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J 1999; 18: 5609–5621.
Tell G, Pellizzari L, Cimarosti D, Pucillo C, Damante G . Ref-1 controls pax-8 DNA-binding activity. Biochem Biophys Res Commun 1998; 252: 178–183.
Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T . Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J 1992; 11: 3323–3335.
Tell G, Quadrifoglio F, Tiribelli C, Kelley MR . The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid Redox Signal 2009; 11: 601–619.
Opresko PL, Cheng WH, von Kobbe C, Harrigan JA, Bohr VA . Werner syndrome and the function of the Werner protein; what they can teach us about the molecular aging process. Carcinogenesis 2003; 24: 791–802.
Grillo C, D’Ambrosio C, Scaloni A, Maceroni M, Merluzzi S, Turano C et al. Cooperative activity of Ref-1/APE and ERp57 in reductive activation of transcription factors. Free Radic Biol Med 2006; 41: 1113–1123.
Chattopadhyay R, Das S, Maiti AK, Boldogh I, Xie J, Hazra TK et al. Regulatory role of human AP-endonuclease (APE1/Ref-1) in YB-1-mediated activation of the multidrug resistance gene MDR1. Mol Cell Biol 2008; 28: 7066–7080.
Sengupta S, Mantha AK, Mitra S, Bhakat KK . Human AP endonuclease (APE1/Ref-1) and its acetylation regulate YB-1-p300 recruitment and RNA polymerase II loading in the drug-induced activation of multidrug resistance gene MDR1. Oncogene 2011; 30: 482–493.
Mantha AK, Dhiman M, Taglialatela G, Perez-Polo RJ, Mitra S . Proteomic study of amyloid beta (25–35) peptide exposure to neuronal cells: impact on APE1/Ref-1’s protein–protein interaction. J Neurosci Res 2012; 90: 1230–1239.
Davydov V, Hansen LA, Shackelford DA . Is DNA repair compromised in Alzheimer’s disease? Neurobiol Aging 2003; 24: 953–968.
Huang E, Qu D, Zhang Y, Venderova K, Haque ME, Rousseaux MWC et al. The role of Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation in neuronal death. Nat Cell Biol 2010; 12: 563–571.
Shaikh AY, Martin LJ . DNA base-excision repair enzyme apurinic/apyrimidinic endonuclease/redox factor-1 is increased and competent in the brain and spinal cord of individuals with amyotrophic lateral sclerosis. Neuromolecular Med 2002; 2: 47–60.
Tan Z, Shi L, Schreiber SS . Differential expression of redox factor-1 associated with beta-amyloid-mediated neurotoxicity. Open Neurosci J 2009; 3: 26–34.
Song SH, Cho EJ, Park MS, Lee YR, Joo HK, Kang G et al. Redox regulating protein APE1/Ref-1 expression is increased in abdominal aortic coarctation-induced hypertension rats. J Korean Soc Hypertens 2012; 18: 126–135.
Al-Attar A, Gossage L, Fareed KR, Shehata M, Mohammed M, Zaitoun AM et al. Human apurinic/apyrimidinic endonuclease (APE1) is a prognostic factor in ovarian, gastro-oesophageal and pancreatico-biliary cancers. Br J Cancer 2010; 102: 704–709.
Di Maso V, Avellini C, Croce LS, Rosso N, Quadrifoglio F, Cesaratto L et al. Subcellular localization of APE1/Ref-1 in human hepatocellular carcinoma: possible prognostic significance. Mol Med 2007; 13: 89–96.
Evans AR, Limp-Foster M, Kelley MR, Going APE . over ref-1. Mutat Res 2000; 461: 83–108.
Herring CJ, West CM, Wilks DP, Davidson SE, Hunter RD, Berry P et al. Levels of the DNA repair enzyme human apurinic/apyrimidinic endonuclease (APE1, APEX, Ref-1) are associated with the intrinsic radiosensitivity of cervical cancers. Br J Cancer 1998; 78: 1128–1133.
Puglisi F, Aprile G, Minisini AM, Barbone F, Cataldi P, Tells G et al. Prognostic significance of Ape1/ref-1 subcellular localization in non-small cell lung carcinomas. Anticancer Res 2001; 21: 4041–4049.
Yoo DG, Song YJ, Cho EJ, Lee SK, Park JB, Yu JH et al. Alteration of APE1/ref-1 expression in non-small cell lung cancer: the implications of impaired extracellular superoxide dismutase and catalase antioxidant systems. Lung Cancer 2008; 60: 277–284.
Duarte DB, Vasko MR . The role of DNA damage and repair in neurotoxicity caused by cancer therapies. In: Kelley MR, (ed).. DNA Repair in Cancer Therapy. Academic Press: San Diego, CA, USA, 2012 pp 283–299.
Englander EW . DNA damage response in peripheral nervous system: coping with cancer therapy-induced DNA lesions. DNA Repair 2013; 12: 685–690.
Mantha AK . APE1: a molecule of focus with neuroprotective and anti-cancer properties. J Biotechnol Biomater 2013; 3: e120.
Chen DS, Herman T, Demple B . Two distinct human DNA diesterases that hydrolyze 3′-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res 1991; 19: 5907–5914.
Fritz G . Human APE/Ref-1 protein. Int J Biochem Cell Biol 2000; 32: 925–929.
Maher RL, Bloom LB . Pre-steady-state kinetic characterization of the AP endonuclease activity of human AP endonuclease 1. J Biol Chem 2007; 282: 30577–30585.
Kanazhevskaya LY, Koval VV, Zharkov DO, Strauss PR, Fedorova OS . Conformational transitions in human AP endonuclease 1 and its active site mutant during abasic site repair. Biochemistry 2010; 49: 6451–6461.
Mol CD, Izumi T, Mitra S, Tainer JA . DNA-bound structures and mutants reveal abasic DNA binding by APE1 DNA repair and coordination. Nature 2000; 403: 451–456.
Jackson EB, Theriot CA, Chattopadhyay R, Mitra S, Izumi T . Analysis of nuclear transport signals in the human apurinic/apyrimidinic endonuclease (APE1/Ref1). Nucleic Acids Res 2005; 33: 3303–3312.
Bhakat KK, Izumi T, Yang SH, Hazra TK, Mitra S . Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J 2003; 22: 6299–6309.
Nunomura A, Moreira PI, Takeda A, Smith MA, Perry G . Oxidative RNA damage and neurodegeneration. Curr Med Chem 2007; 14: 2968–2975.
Barnes T, Kim WC, Mantha AK, Kim SE, Izumi T, Mitra S et al. Identification of Apurinic/apyrimidinic endonuclease 1 (APE1) as the endoribonuclease that cleaves c-myc mRNA. Nucleic Acids Res 2009; 37: 3946–3958.
Tell G, Wilson DM, Lee CH . Intrusion of a DNA repair protein in the RNome world: is this the beginning of a new era? Mol Cell Biol 2010; 30: 366–371.
Slupphaug G, Kavli B, Krokan HE . The interacting pathways for prevention and repair of oxidative DNA damage. Mutat Res 2003; 531: 231–251.
Hegde ML, Mantha AK, Hazra TK, Bhakat KK, Mitra S, Szczesny B . Oxidative genome damage and its repair: implications in aging and neurodegenerative diseases. Mech Ageing Dev 2012; 133: 157–168.
Demple B, Harrison L . Repair of oxidative damage to DNA: enzymology and biology. Annu Rev Biochem 1994; 63: 915–948.
Doetsch PW, Cunningham RP . The enzymology of apurinic/apyrimidinic endonucleases. Mutat Res 1990; 236: 173–201.
Fung H, Demple B . A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol Cell 2005; 17: 463–470.
Sobol RW, Wilson SH . Mammalian DNA beta-polymerase in base excision repair of alkylation damage. Prog Nucleic Acid Res Mol Biol 2001; 68: 57–74.
Krokan HE, Standal R, Slupphaug G . DNA glycosylases in the base excision repair of DNA. Biochem J 1997; 325 (Pt 1): 1–16.
Hazra TK, Izumi T, Kow YW, Mitra S . The discovery of a new family of mammalian enzymes for repair of oxidatively damaged DNA, and its physiological implications. Carcinogenesis 2003; 24: 155–157.
Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD et al. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 2001; 104: 107–117.
Izumi T, Hazra TK, Boldogh I, Tomkinson AE, Park MS, Ikeda S et al. Requirement for human AP endonuclease 1 for repair of 3′-blocking damage at DNA single-strand breaks induced by reactive oxygen species. Carcinogenesis 2000; 21: 1329–1334.
Loeb LA, Preston BD . Mutagenesis by apurinic/apyrimidinic sites. Annu Rev Genet 1986; 20: 201–230.
Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M et al. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell 2004; 15: 209–220.
Hazra TK, Kow YW, Hatahet Z, Imhoff B, Boldogh I, Mokkapati SK et al. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J Biol Chem 2002; 277: 30417–30420.
Abate C, Patel L, Rauscher FJ, Curran T . Redox regulation of fos and jun DNA-binding activity in vitro. Science 1990; 249: 1157–1161.
Luo M, Zhang J, He H, Su D, Chen Q, Gross ML et al. Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease. Biochemistry 2012; 51: 695–705.
Walker LJ, Robson CN, Black E, Gillespie D, Hickson ID . Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol Cell Biol 1993; 13: 5370–5376.
Gorman MA, Morera S, Rothwell DG, de La Fortelle E, Mol CD, Tainer JA et al. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J 1997; 16: 6548–6558.
Qin J, Clore GM, Kennedy WP, Kuszewski J, Gronenborn AM . The solution structure of human thioredoxin complexed with its target from Ref-1 reveals peptide chain reversal. Structure 1996; 4: 613–620.
Seo YR, Kelley MR, Smith ML . Selenomethionine regulation of p53 by a ref1-dependent redox mechanism. Proc Natl Acad Sci USA 2002; 99: 14548–14553.
Vasko MR, Guo C, Kelley MR . The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA Repair 2005; 4: 367–379.
Jeon BH, Gupta G, Park YC, Qi B, Haile A, Khanday FA et al. Apurinic/apyrimidinic endonuclease 1 regulates endothelial NO production and vascular tone. Circ Res 2004; 95: 902–910.
Pines A, Bivi N, Romanello M, Damante G, Kelley MR, Adamson ED et al. Cross-regulation between Egr-1 and APE/Ref-1 during early response to oxidative stress in the human osteoblastic HOBIT cell line: evidence for an autoregulatory loop. Free Radic Res 2005; 39: 269–281.
Zou GM, Luo MH, Reed A, Kelley MR, Yoder MC . Ape1 regulates hematopoietic differentiation of embryonic stem cells through its redox functional domain. Blood 2007; 109: 1917–1922.
Ordway JM, Eberhart D, Curran T . Cysteine 64 of Ref-1 is not essential for redox regulation of AP-1 DNA binding. Mol Cell Biol 2003; 23: 4257–4266.
Ando K, Hirao S, Kabe Y, Ogura Y, Sato I, Yamaguchi Y et al. A new APE1/Ref-1-dependent pathway leading to reduction of NF-{kappa}B and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res 2008; 36: 4327–4336.
Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M et al. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxid Redox Signal 2008; 10: 1853–1867.
Georgiadis MM, Luo M, Gaur RK, Delaplane S, Li X, Kelley MR . Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat Res 2008; 643: 54–63.
Vascotto C, Bisetto E, Li M, Zeef LA, D’Ambrosio C, Domenis R et al. Knock-in reconstitution studies reveal an unexpected role of Cys-65 in regulating APE1/Ref-1 subcellular trafficking and function. Mol Biol Cell 2011; 22: 3887–3901.
Shimizu N, Sugimoto K, Tang J, Nishi T, Sato I, Hiramoto M et al. High-performance affinity beads for identifying drug receptors. Nat Biotechnol 2000; 18: 877–881.
Zhang J, Luo M, Marasco D, Logsdon D, LaFavers KA, Chen Q et al. Inhibition of apurinic/apyrimidinic endonuclease I's redox activity revisited. Biochemistry 2013; 52: 2955–2966.
Su D, Delaplane S, Luo M, Rempel DL, Vu B, Kelley MR et al. Interactions of apurinic/apyrimidinic endonuclease with a redox inhibitor: evidence for an alternate conformation of the enzyme. Biochemistry 2011; 50: 82–92.
Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J . AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci USA 1997; 94: 3633–3638.
Guan Z, Basi D, Li Q, Mariash A, Xia YF, Geng JG et al. Loss of redox factor 1 decreases NF-κB activity and increases susceptibility of endothelial cells to apoptosis. Arterioscler Thromb Vasc Biol 2005; 25: 96–101.
Jayaraman L, Murthy KG, Zhu C, Curran T, Xanthoudakis S, Prives C . Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev 1997; 11: 558–570.
Hsieh MM, Hegde V, Kelley MR, Deutsch WA . Activation of APE/Ref-1 redox activity is mediated by reactive oxygen species and PKC phosphorylation. Nucleic Acids Res 2001; 29: 3116–3122.
Ramana CV, Boldogh I, Izumi T, Mitra S . Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc Natl Acad Sci USA 1998; 95: 5061–5066.
Suzuki S, Nagaya T, Suganuma N, Tomoda Y, Seo H . Inductions of immediate early genes (IEGs) and ref-1 by human chorionic gonadotropin in murine Leydig cell line (MA-10). IUBMB Life 1998; 44: 217–224.
Tell G, Damante G, Caldwell D, Kelley MR . The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid Redox Signal 2005; 7: 367–384.
Seet BT, Dikic I, Zhou MM, Pawson T . Reading protein modifications with interaction domains. Nat Rev Mol Cell Biol 2006; 7: 473–483.
Fantini D, Vascotto C, Marasco D, D’Ambrosio C, Romanello M, Vitagliano L et al. Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions. Nucleic Acids Res 2010; 38: 8239–8256.
Lirussi L, Antoniali G, Vascotto C, D’Ambrosio C, Poletto M, Romanello M et al. Nucleolar accumulation of APE1 depends on charged lysine residues that undergo acetylation upon genotoxic stress and modulate its BER activity in cells. Mol Biol Cell 2012; 23: 4079–4096.
Vascotto C, Fantini D, Romanello M, Cesaratto L, Deganuto M, Leonardi A et al. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol Cell Biol 2009; 29: 1834–1854.
Busso CS, Iwakuma T, Izumi T . Ubiquitination of mammalian AP endonuclease (APE1) regulated by the p53–MDM2 signaling pathway. Oncogene 2009; 28: 1616–1625.
Yacoub A, Kelley MR, Deutsch WA . The DNA repair activity of human redox/repair protein APE/Ref-1 is inactivated by phosphorylation. Cancer Res 1997; 57: 5457–5459.
Fritz G, Kaina B . Phosphorylation of the DNA repair protein APE/REF-1 by CKII affects redox regulation of AP-1. Oncogene 1999; 18: 1033–1040.
Parsons JL, Tait PS, Finch D, Dianova II, Edelmann MJ, Khoronenkova SV et al. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J 2009; 28: 3207–3215.
Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 1989; 243: 1576–1583.
Busso CS, Lake MW, Izumi T . Posttranslational modification of mammalian AP endonuclease (APE1). Cell Mol Life Sci 2010; 67: 3609–3620.
Nijman S, Luna-Vargas M, Velds A, Brummelkamp TR, Dirac AMG, Sixma TK et al. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005; 123: 773–786.
Busso CS, Wedgeworth CM, Izumi T . Ubiquitination of human AP-endonuclease 1 (APE1) enhanced by T233E substitution and by CDK5. Nucleic Acids Res 2011; 39: 8017–8028.
Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K et al. Distinct roles of thioredoxin in the cytoplasm and in the nucleus a two-step mechanism of redox regulation of transcription factor NF-κB. J Biol Chem 1999; 274: 27891–27897.
Wei SJ, Botero A, Hirota K, Bradbury CM, Markovina S, Laszlo A et al. Thioredoxin nuclear translocation and interaction with redox factor-1 activates the activator protein-1 transcription factor in response to ionizing radiation. Cancer Res 2000; 60: 6688–6695.
Wiesel P, Foster LC, Pellacani A, Layne MD, Hsieh CM, Huggins GS et al. Thioredoxin facilitates the induction of heme oxygenase-1 in response to inflammatory mediators. J Biol Chem 2000; 275: 24840–24846.
Wang YT, Tzeng DW, Wang CY, Hong JY, Yang JL . APE1/Ref-1 prevents oxidative inactivation of ERK for G1-to-S progression following lead acetate exposure. Toxicology 2013; 305: 120–129.
Ames BN, Shigenaga MK, Hagen TM . Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA 1993; 90: 7915–7922.
Aruoma OI, Kaur H, Halliwell B . Oxygen free radicals and human diseases. J R Soc Promot Health 1991; 111: 172–177.
Dedon PC, Tannenbaum SR . Reactive nitrogen species in the chemical biology of inflammation. Arch Biochem Biophys 2004; 423: 12–22.
Inoue M, Sato EF, Nishikawa M, Park AM, Kira Y, Imada I et al. Mitochondrial generation of reactive oxygen species and its role in aerobic life. Curr Med Chem 2003; 10: 2495–2505.
Turrens JF . Mitochondrial formation of reactive oxygen species. J Physiol 2003; 552: 335–344.
Martin KR, Barrett JC . Reactive oxygen species as double-edged swords in cellular processes: low-dose cell signaling versus high-dose toxicity. Hum Exp Toxicol 2002; 21: 71–75.
Halliwell B . Reactive oxygen species in living systems: source, biochemistry, and role in human disease. Am J Med 1991; 91: S14–S22.
Valko M, Morris H, Cronin MTD. Metals . toxicity and oxidative stress. Curr Med Chem 2005; 12: 1161–1208.
Trachootham D, Lu W, Ogasawara MA, Valle NRD, Huang P . Redox regulation of cell survival. Antioxid Redox Signal 2008; 10: 1343–1374.
Smith DG, Cappai R, Barnham KJ . The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochim Biophys Acta 2007; 1768: 1976–1990.
Mitra S, Boldogh I, Izumi T, Hazra TK . Complexities of the DNA base excision repair pathway for repair of oxidative DNA damage. Environ Mol Mutagen 2001; 38: 180–190.
Silber JR, Bobola MS, Blank A, Schoeler KD, Haroldson PD, Huynh MB et al. The apurinic/apyrimidinic endonuclease activity of Ape1/Ref-1 contributes to human glioma cell resistance to alkylating agents and is elevated by oxidative stress. Clin Cancer Res 2002; 8: 3008–3018.
Angkeow P, Deshpande SS, Qi B, Liu YX, Park YC, Jeon BH et al. Redox factor-1: an extra-nuclear role in the regulation of endothelial oxidative stress and apoptosis. Cell Death Differ 2002; 9: 717–725.
Hafsi H, Hainaut P . Redox control and interplay between p53 isoforms: roles in the regulation of basal p53 levels, cell fate, and senescence. Antioxid Redox Signal 2011; 15: 1655–1667.
Maillet A, Pervaiz S . Redox regulation of p53, redox effectors regulated by p53: a subtle balance. Antioxid Redox Signal 2012; 16: 1285–1294.
Ozaki M, Suzuki S, Irani K . Redox factor-1/APE suppresses oxidative stress by inhibiting the rac1 GTPase. FASEB J 2002; 16: 889–890.
Kenny MK, Mendez F, Sandigursky M, Kureekattil RP, Goldman JD, Franklin WA et al. Heat shock protein 70 binds to human apurinic/apyrimidinic endonuclease and stimulates endonuclease activity at abasic sites. J Biol Chem 2001; 276: 9532–9536.
El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW . A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res 2003; 31: 5526–5533.
Qin J, Clore GM, Kennedy WM, Huth JR, Gronenborn AM . Solution structure of human thioredoxin in a mixed disulfide intermediate complex with its target peptide from the transcription factor NFκB. Structure 1995; 3: 289–297.
Gray MJ, Zhang J, Ellis LM, Semenza GL, Evans DB, Watowich SS et al. HIF-1α, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene 2005; 24: 3110–3120.
Hanson S, Kim E, Deppert W . Redox factor 1 (Ref-1) enhances specific DNA binding of p53 by promoting p53 tetramerization. Oncogene 2005; 24: 1641–1647.
Ray S, Lee C, Hou T, Bhakat KK, Brasier AR . Regulation of signal transducer and activator of transcription 3 enhanceosome formation by apurinic/apyrimidinic endonuclease 1 in hepatic acute phase response. Mol Endocrinol 2010; 24: 391–401.
Sies H . Strategies of antioxidant defense. Eur J Biochem 1994; 215: 213–219.
Sies H . Oxidative stress: oxidants and antioxidants. Exp Physiol 1997; 82: 291–295.
Kohen R, Nyska A . Invited review: oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol Pathol 2002; 30: 620–650.
Kim CS, Son SJ, Kim EK, Kim SN, Yoo DG, Kim HS et al. Apurinic/apyrimidinic endonuclease1/redox factor-1 inhibits monocyte adhesion in endothelial cells. Cardiovasc Res 2006; 69: 520–526.
Hussain SP, Amstad P, He P, Robles A, Lupold S, Kaneko I et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res 2004; 64: 2350–2356.
Zaky A, Busso C, Izumi T, Chattopadhyay R, Bassiouny A, Mitra S et al. Regulation of the human AP-endonuclease (APE1/Ref-1) expression by the tumor suppressor p53 in response to DNA damage. Nucleic Acids Res 2008; 36: 1555–1566.
Zitomer RS, Lowry CV . Regulation of gene expression by oxygen in Saccharomyces cerevisiae. Microbiol Rev 1992; 56: 1–11.
Chen S, Xiong GS, Wu S, Mo J . Downregulation of apurinic/apyrimidinic endonuclease 1/redox factor-1 enhances the sensitivity of human pancreatic cancer cells to radiotherapy in vitro. Cancer Biother Radiopharm 2012; 28: 169–176.
Naganuma T, Nakayama T, Sato N, Fu Z, Soma M, Yamaguchi M et al. Haplotype-based case–control study on human apurinic/apyrimidinic endonuclease 1/redox effector factor-1 gene and essential hypertension. Am J Hypertens 2009; 23: 186–191.
Marcon G, Tell G, Perrone L, Garbelli R, Quadrifoglio F, Tagliavini F et al. APE1/Ref-1 in Alzheimer’s disease: an immunohistochemical study. Neurosci Lett 2009; 466: 124–127.
Gencer M, Dasdemir S, Cakmakoglu B, Cetinkaya Y, Varlibas F, Tireli H et al. DNA repair genes in Parkinson’s disease. Genet Test Mol Biomarkers 2012; 16: 504–507.
Siddiqui A, Rivera-Sanchez S, Castro MR, Acevedo-Torres K, Rane A, Torres-Ramos CA et al. Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington’s disease. Free Radic Biol Med 2012; 53: 1478–1488.
Coppede F . An overview of DNA repair in amyotrophic lateral sclerosis. Scientific World J 2011; 11: 1679–1691.
Stetler RA, Gao Y, Zukin RS, Vosler PS, Zhang L, Zhang F et al. Apurinic/apyrimidinic endonuclease APE1 is required for PACAP-induced neuroprotection against global cerebral ischemia. Proc Natl Acad Sci USA 2010; 107: 3204–3209.
Yan N, Cherepanov P, Daigle JE, Engelman A, Lieberman J . The SET complex acts as a barrier to autointegration of HIV-1. PLoS Pathog 2009; 5: e1000327.
Yang JL, Tadokoro T, Keijzers G, Mattson MP, Bohr VA . Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREB-mediated up-regulation of apurinic endonuclease 1. J Biol Chem 2010; 285: 28191–28199.
Lewandowski NM, Bordelon Y, Brickman AM, Angulo S, Khan U, Muraskin J et al. Regional vulnerability in Huntington's disease: fMRI-guided molecular analysis in patients and a mouse model of disease. Neurobiol Dis 2012; 52: 84–93.
Rothstein JD . Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol 2009; 65: S3–S9.
Kisby GE, Milne J, Sweatt C . Evidence of reduced DNA repair in amyotrophic lateral sclerosis brain tissue. Neuroreport 1997; 8: 1337–1339.
Hadi MZ, Coleman MA, Fidelis K, Mohrenweiser HW, Wilson DM III . Functional characterization of Ape1 variants identified in the human population. Nucleic Acids Res 2000; 28: 3871–3879.
Hayward C, Colville S, Swingler RJ, Brock DJH . Molecular genetic analysis of the APEX nuclease gene in amyotrophic lateral sclerosis. Neurology 1999; 52: 1899–1899.
Liu PK . DNA damage and repair in the brain after cerebral ischemia. Curr Top Med Chem 2001; 1: 483–495.
Edwards M, Kent TA, Rea HC, Wei J, Quast M, Izumi T et al. APE/Ref-1 responses to ischemia in rat brain. Neuroreport 1998; 9: 4015–4018.
Morita-Fujimura Y, Fujimura M, Kawase M, Chan PH . Early decrease in apurinic/apyrimidinic endonuclease is followed by DNA fragmentation after cold injury-induced brain trauma in mice. Neuroscience 1999; 93: 1465–1473.
Bertram JS, Kolonel LN, Meyskens FL . Rationale and strategies for chemoprevention of cancer in humans. Cancer Res 1987; 47: 3012–3031.
Luo M, Kelley MR . Inhibition of the human apurinic/apyrimidinic endonuclease (APE1) repair activity and sensitization of breast cancer cells to DNA alkylating agents with lucanthone. Anticancer Res 2004; 24: 2127–2134.
Yang S, Irani K, Heffron SE, Jurnak F, Meyskens FL . Alterations in the expression of the apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE/Ref-1) in human melanoma and identification of the therapeutic potential of resveratrol as an APE/Ref-1 inhibitor. Mol Cancer Ther 2005; 4: 1923–1935.
Bobola MS, Blank A, Berger MS, Stevens BA, Silber JR . Apurinic/apyrimidinic endonuclease activity is elevated in human adult gliomas. Clin Cancer Res 2001; 7: 3510–3518.
Wang D, Luo M, Kelley MR . Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol Cancer Ther 2004; 3: 679–686.
Koukourakis MI, Giatromanolaki A, Kakolyris S, Sivridis E, Georgoulias V, Funtzilas G et al. Nuclear expression of human apurinic/apyrimidinic endonuclease (HAP1/Ref-1) in head-and-neck cancer is associated with resistance to chemoradiotherapy and poor outcome. Int J Radiat Oncol Biol Phys 2001; 50: 27–36.
Sak SC, Harnden P, Johnston CF, Paul AB, Kiltie AE . APE1 and XRCC1 protein expression levels predict cancer-specific survival following radical radiotherapy in bladder cancer. Clin Cancer Res 2005; 11: 6205–6211.
Thomson B, Tritt R, Davis M, Kelley MR . Histology-specific expression of a DNA repair protein in pediatric rhabdomyosarcomas. J Pediatr Hematol Oncol 2001; 23: 234–239.
Tell G, Zecca A, Pellizzari L, Spessotto P, Colombatti A, Kelley MR et al. An ‘environment to nucleus’ signaling system operates in B lymphocytes: redox status modulates BSAP/Pax-5 activation through Ref-1 nuclear translocation. Nucleic Acids Res 2000; 28: 1099–1105.
Bobola MS, Finn LS, Ellenbogen RG, Geyer JR, Berger MS, Braga JM et al. Apurinic/apyrimidinic endonuclease activity is associated with response to radiation and chemotherapy in medulloblastoma and primitive neuroectodermal tumors. Clin Cancer Res 2005; 11: 7405–7414.
Cardoso AA, Jiang Y, Luo M, Reed AM, Shahda S, He Y et al. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS ONE 2012; 7: e47462.
Fishel ML, Jiang Y, Rajeshkumar NV, Scandura G, Sinn AL, He Y et al. Impact of APE1/Ref-1 redox inhibition on pancreatic tumor growth. Mol Cancer Ther 2011; 10: 1698–1708.
Cun Y, Dai N, Xiong C, Li M, Sui J, Qian C et al. Silencing of APE1 enhances sensitivity of human hepatocellular carcinoma cells to radiotherapy in vitro and in a xenograft model. PLoS ONE 2013; 8: e55313.
Kelley MR, Fishel ML . DNA repair proteins as molecular targets for cancer therapeutics. Anticancer Agents Med Chem 2008; 8: 417–425.
Bapat A, Glass LS, Luo M, Fishel ML, Long EC, Georgiadis MM et al. Novel small-molecule inhibitor of apurinic/apyrimidinic endonuclease 1 blocks proliferation and reduces viability of glioblastoma cells. J Pharmacol Exp Ther 2010; 334: 988–998.
Kim MH, Kim HB, Acharya S, Sohn HM, Jun JY, Chang IY et al. Ape1/Ref-1 induces glial cell-derived neurotropic factor (GDNF) responsiveness by upregulating GDNF receptor α1 expression. Mol Cell Biol 2009; 29: 2264–2277.
Luo M, Caldwell D, Xu Y, He Y, Reed A, Handa H et al. Inhibition of the human apurinic/apyrimidinic endonuclease DNA base excision repair enzyme/redox factor (APE1/Ref-1) using small molecule redox and repair inhibitors: therapeutic implications [abstract]. Proc Amer Assoc Cancer Res (AACR Meeting Abstracts) 2004; 45 Abstract nr 3042.
Canbay E, Agachan B, Gulluoglu M, Isbir T, Balik E, Yamaner S et al. Possible associations of APE1 polymorphism with susceptibility and HOGG1 polymorphism with prognosis in gastric cancer. Anticancer Res 2010; 30: 1359–1364.
Chen L, Ambrosone CB, Lee J, Sellers TA, Pow-Sang J, Park JY . Association between polymorphisms in the DNA repair genes XRCC1 and APE1, and the risk of prostate cancer in White and Black Americans. J Urol 2006; 175: 108–112.
Lin CH, Chen PM, Cheng YW, Chen CY, Yuan CJ, Lee H . The APE1 Asp/Asp genotype and the combination of APE1 Asp/Asp and hOGG1-Cys variants are associated with increased p53 mutation in non–small cell lung cancer. J Epidemiol 2012; 22: 537–542.
Liu C, Yin Q, Li L, Zhuang Y, Zu X, Wang Y . APE1 Asp148Glu gene polymorphism and bladder cancer risk: a meta-analysis. Mol Biol Rep 2013; 40: 171–176.
Pieretti M, Khattar NH, Smith SA . Common polymorphisms and somatic mutations in human base excision repair genes in ovarian and endometrial cancers. Mutat Res 2001; 432: 53–59.
Shinmura K, Yamaguchi S, Saitoh T, Kohno T, Yokota J . Somatic mutations and single nucleotide polymorphisms of base excision repair genes involved in the repair of 8-hydroxyguanine in damaged DNA. Cancer Lett 2001; 166: 65–69.
Lo YL, Jou YS, Hsiao CF, Chang GC, Tsai YH, Su WC et al. A polymorphism in the APE1 gene promoter is associated with lung cancer risk. Cancer Epidemiol Biomarkers Prev 2009; 18: 223–229.
Cao Q, Qin C, Meng X, Ju X, Ding Q, Wang M et al. Genetic polymorphisms in APE1 are associated with renal cell carcinoma risk in a Chinese population. Mol Carcinog 2011; 50: 863–870.
Cervelli T, Borghini A, Galli A, Andreassi MG . DNA damage and repair in atherosclerosis: current insights and future perspectives. Int J Mol Sci 2012; 13: 16929–16944.
Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB et al. Executive summary: heart disease and stroke statistics—2012 update a report from the American Heart Association. Circulation 2012; 125: 188–197.
Patterson C . Blood pressure control goes nuclear. Circ Res 2004; 95: 849–851.
Gokkusu C, Cakmakoglu B, Dasdemir S, Tulubas F, Elitok A, Tamer S et al. Association between genetic variants of DNA repair genes and coronary artery disease. Genet Test Mol Biomarkers 2013; 17: 307–313.
Sengupta S, Chattopadhyay R, Mantha AK, Mitra S, Bhakat KK . Regulation of mouse-renin gene by apurinic/apyrimidinic-endonuclease 1 (APE1/Ref-1) via recruitment of histone deacetylase 1 corepressor complex. J Hypertens 2012; 30: 917–925.
O’Hara AM, Bhattacharyya A, Mifflin RC, Smith MF, Ryan KA, Scott KGE et al. Interleukin-8 induction by Helicobacter pylori in gastric epithelial cells is dependent on apurinic/apyrimidinic endonuclease-1/redox factor-1. J Immunol 2006; 177: 7990–7999.
Bhattaracharyya A, Chattopadhyay R, Burnette BR, Cross JV, Mitra S, Ernst PB et al. Acetylation of apurinic/apyrimidinic endonuclease-1 regulates Helicobacter pylori-mediated gastric epithelial cell apoptosis. Gastroenterology 2009; 136: 2258–2269.
Chattopadhyay R, Bhattacharyya A, Crowe SE . Dual regulation by apurinic/apyrimidinic endonuclease-1 inhibits gastric epithelial cell apoptosis during Helicobacter pylori infection. Cancer Res 2010; 70: 2799–2808.
Jiang A, Gao H, Kelley MR, Qiao X . Inhibition of APE1/Ref-1 redox activity with APX3330 blocks retinal angiogenesis in vitro and in vivo. Vision Res 2011; 51: 93–100.
Chiarini LB, Linden R . Tissue biology of apoptosis. Ref-1 and cell differentiation in the developing retina. Ann N Y Acad Sci 2000; 926: 64–78.
Raffoul JJ, Banerjee S, Singh-Gupta V, Knoll Z, Fite A, Zhang H et al. Down-regulation of apurinic/apyrimidinic endonuclease 1/redox factor-1 expression by soy isoflavones enhances prostate cancer radiotherapy in vitro and in vivo. Cancer Res 2007; 67: 2141–2149.
Kingsley K, Truong K, Low E, Hill CK, Chokshi SB, Phipps D et al. Soy Protein extract (SPE) exhibits differential in vitro cell proliferation effects in oral cancer and normal cell lines. J Diet Suppl 2011; 8: 169–188.
Singh-Gupta V, Joiner MC, Runyan L, Yunker CK, Sarkar FH, Miller S et al. Soy isoflavones augment radiation effect by inhibiting APE1/Ref-1 DNA repair activity in non-small cell lung cancer. J Thorac Oncol 2011; 6: 688–698.
Singh-Gupta V, Zhang H, Banerjee S, Kong D, Raffoul JJ, Sarkar FH et al. Radiation induced HIF-1α cell survival pathway is inhibited by soy isoflavones in prostate cancer cells. Int J Cancer 2009; 124: 1675–1684.
Singh-Gupta V, Zhang H, Yunker CK, Ahmad Z, Zwier D, Sarkar FH et al. Daidzein effect on hormone refractory prostate cancer in vitro and in vivo compared to genistein and soy extract: potentiation of radiotherapy. Pharm Res 2010; 27: 1115–1127.
Wang Y, Raffoul JJ, Che M, Doerge DR, Joiner MC, Kucuk O et al. Prostate cancer treatment is enhanced by genistein in vitro and in vivo in a syngeneic orthotopic tumor model. Radiat Res 2006; 166: 73–80.
Sarkaki A, Amani R, Badavi M, Moghaddam AZ, Aligholi H, Safahani M et al. Pre-treatment effect of different doses of soy isoflavones on spatial learning and memory in an ovariectomized animal model of Alzheimer’s disease. Pak J Biol Sci 2008; 11: 1114–1119.
Hsieh HM, Wu WM, Hu ML . Soy isoflavones attenuate oxidative stress and improve parameters related to aging and Alzheimer’s disease in C57BL/6J mice treated with D-galactose. Food Chem Toxicol 2009; 47: 625–632.
Ho KP, Li L, Zhao L, Qian ZM . Genistein protects primary cortical neurons from iron-induced lipid peroxidation. Mol Cell Biochem 2003; 247: 219–222.
Lee YB, Lee HJ, Sohn HS . Soy isoflavones and cognitive function. J Nutr Biochem 2005; 16: 641–649.
Gao X, Xu YX, Divine G, Janakiraman N, Chapman RA, Gautam SC . Disparate in vitro and in vivo antileukemic effects of resveratrol, a natural polyphenolic compound found in grapes. J Nutr 2002; 132: 2076–2081.
Wong DH, Villanueva JA, Cress AB, Sokalska A, Ortega I, Duleba AJ . Resveratrol inhibits the mevalonate pathway and potentiates the antiproliferative effects of simvastatin in rat theca-interstitial cells. Fertil Steril 2011; 96: 1252–1258.
Zaky A, Mohammad B, Moftah M, Kandeel KM, Bassiouny AR . Apurinic/apyrimidinic endonuclease 1 is a key modulator of aluminum-induced neuroinflammation. BMC Neurosci 2013; 14: 1–12.
Petrovski G, Gurusamy N, Das DK . Resveratrol in cardiovascular health and disease. Ann N Y Acad Sci 2011; 1215: 22–33.
Gurusamy N, Ray D, Lekli I, Das DK . Red wine antioxidant resveratrol-modified cardiac stem cells regenerate infarcted myocardium. J Cell Mol Med 2010; 14: 2235–2239.
Liu Z, Song Y, Zhang X, Liu Z, Zhang W, Mao W et al. Effects of trans-resveratrol on hypertension-induced cardiac hypertrophy using the partially nephrectomized rat model. Clin Exp Pharmacol Physiol 2005; 32: 1049–1054.
Juric D, Wojciechowski P, Das DK, Netticadan T . Prevention of concentric hypertrophy and diastolic impairment in aortic-banded rats treated with resveratrol. Am J Physiol Heart Circ Physiol 2007; 292: H2138–H2143.
Kunchandy E, Rao MNA . Oxygen radical scavenging activity of curcumin. Int J Pharm 1990; 58: 237–240.
Aggarwal BB, Kumar A, Bharti AC . Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res 2003; 23: 363–398.
Rao CV, Rivenson A, Simi B, Reddy BS . Chemoprevention of colon carcinogenesis by dietary curcumin, a naturally occurring plant phenolic compound. Cancer Res 1995; 55: 259–266.
Mukhopadhyay A, Bueso-Ramos C, Chatterjee D, Pantazis P, Aggarwal BB . Curcumin downregulates cell survival mechanisms in human prostate cancer cell lines. Oncogene 2001; 20: 7597–7609.
Moragoda L, Jaszewski R, Majumdar AP . Curcumin induced modulation of cell cycle and apoptosis in gastric and colon cancer cells. Anticancer Res 2001; 21: 873–878.
Cheng AL, Hsu CH, Jen-K Lin, Hsu MM, Ho YF, Shen TS et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res 2001; 21: 2895–2900.
Chen YR, Tan TH . Inhibition of the c-Jun N-terminal kinase (JNK) signaling pathway by curcumin. Oncogene 1998; 17: 173–178.
Shoba G, Joy D, Joseph T, Majeed M, Rajendran R, Srinivas PSSR . Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med 2007; 64: 353–356.
Scapagnini G, Colombrita C, Amadio M, D’Agata V, Arcelli E, Sapienza M et al. Curcumin activates defensive genes and protects neurons against oxidative stress. Antioxid Redox Signal 2006; 8: 395–403.
Hegde ML, Hegde PM, Holthauzen LMF, Hazra TK, Rao KSJ, Mitra S . Specific inhibition of NEIL-initiated repair of oxidized base damage in human genome by copper and iron potential etiological linkage to neurodegenerative diseases. J Biol Chem 2010; 285: 28812–28825.
Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM . The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci 2001; 21: 8370–8377.
Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 2005; 280: 5892–5901.
Bassiouny AR, Atteya MA, Fatma H, Neenaa HM . Curcumin and EGCG suppress apurinic/apyrimidinic endonuclease 1 and induce complete remission in B-cell non-Hodgkin’s lymphoma patients. Functional Foods in Health Dis 2011; 1: 525–544.
Jin L, Li C, Xu Y, Wang L, Liu J, Wang D et al. Epigallocatechin gallate promotes p53 accumulation and activity via the inhibition of MDM2-mediated p53 ubiquitination in human lung cancer cells. Oncol Rep 2013; 29: 1983–1990.
Singh T, Katiyar SK . Green tea catechins reduce invasive potential of human melanoma cells by targeting COX-2, PGE2 receptors and epithelial-to-mesenchymal transition. PLoS ONE 2011; 6: e25224.
Yim D, Singh RP, Agarwal C, Lee S, Chi H, Agarwal R . A novel anticancer agent, decursin, induces G1 arrest and apoptosis in human prostate carcinoma cells. Cancer Res 2005; 65: 1035–1044.
Dong J, Sulik KK, Chen SY . Nrf2-mediated transcriptional induction of antioxidant response in mouse embryos exposed to ethanol in vivo: implications for the prevention of fetal alcohol spectrum disorders. Antioxid Redox Signal 2008; 10: 2023–2033.
Li L, Li W, Jung SW, Lee YW, Kim YH . Protective effects of decursin and decursinol angelate against amyloid. BETA.-protein-induced oxidative stress in the PC12 cell line: the role of Nrf2 and antioxidant enzymes. Biosci Biotechnol Biochem 2011; 75: 434–442.
Bhat TA, Moon JS, Lee S, Yim D, Singh RP . Inhibition of angiogenic attributes by decursin in endothelial cells and ex vivo rat aortic ring angiogenesis model. Indian J Exp Biol 2011; 49: 848–856.
Kang SY, Lee KY, Park MJ, Kim YC, Markelonis GJ, Oh TH et al. Decursin from Angelica gigas mitigates amnesia induced by scopolamine in mice. Neurobiol Learn Mem 2003; 79: 11–18.
Yang CS, Landau JM, Huang MT, Newmark HL . Inhibition of carcinogenesis by dietary polyphenolic compounds. Annu Rev Nutr 2001; 21: 381–406.
Abarikwu SO, Pant AB, Farombi EO . Dietary antioxidant, quercetin, protects sertoli germ cell coculture from atrazine induced oxidative damage. J Biochem Mol Toxicol 2012; 26: 477–485.
Jordan MJ, Lax V, Rota MC, Loran S, Sotomayor JA . Relevance of carnosic acid, carnosol, and rosmarinic acid concentrations in the in vitro antioxidant and antimicrobial activities of rosmarinus officinalis (L.) methanolic extracts. J Agric Food Chem 2012; 60: 9603–9608.
Petersen M, Simmonds MSJ . Rosmarinic acid. Phytochemistry 2003; 62: 121–125.
Aherne SA, O'Brien NM . Protection by the flavonoids myricetin, quercetin, and rutin against hydrogen peroxide-induced DNA damage in Caco-2 and Hep G2 cells. Nutr Cancer 1999; 34: 160–166.
Horvathova K, Chalupa I, Sebova L, Tothova D, Vachalkova A . Protective effect of quercetin and luteolin in human melanoma HMB-2 cells. Mutat Res 2005; 565: 105–112.
Horvathova K, Novotny L, Vachalkova A . The free radical scavenging activity of four flavonoids determined by the comet assay. Neoplasma 2003; 50: 291–295.
Lima CF, Fernandes-Ferreira M, Pereira-Wilson C . Phenolic compounds protect HepG2 cells from oxidative damage: relevance of glutathione levels. Life Sci 2006; 79: 2056–2068.
Furtado RA, De Araujo FRR, Resende FA, Cunha WR, Tavares DC . Protective effect of rosmarinic acid on V79 cells evaluated by the micronucleus and comet assays. J Appl Toxicol 2010; 30: 254–259.
Silva JP, Gomes AC, Coutinho OP . Oxidative DNA damage protection and repair by polyphenolic compounds in PC12 cells. Eur J Pharmacol 2008; 601: 50–60.
Ramos AA, Azqueta A, Pereira-Wilson C, Collins AR . Polyphenolic compounds from Salvia species protect cellular DNA from oxidation and stimulate DNA repair in cultured human cells. J Agric Food Chem 2010; 58: 7465–7471.
Qu J, Liu GH, Huang B, Chen C . Nitric oxide controls nuclear export of APE1/Ref-1 through S-nitrosation of cysteines 93 and 310. Nucleic Acids Res 2007; 35: 2522–2532.
Lee SK, Chung JI, Park MS, Joo HK, Lee EJ, Cho EJ et al. Apurinic/apyrimidinic endonuclease 1 inhibits protein kinase C-mediated p66shc phosphorylation and vasoconstriction. Cardiovasc Res 2011; 91: 502–509.
Ranalli TA, Tom S, Bambara RA . AP endonuclease 1 coordinates flap endonuclease 1 and DNA ligase I activity in long patch base excision repair. J Biol Chem 2002; 277: 41715–41724.
Sukhanova MV, Khodyreva SN, Lebedeva NA, Prasad R, Wilson SH, Lavrik OI . Human base excision repair enzymes apurinic/apyrimidinic endonuclease1 (APE1), DNA polymerase beta and poly(ADP-ribose) polymerase 1: interplay between strand-displacement DNA synthesis and proofreading exonuclease activity. Nucleic Acids Res 2005; 33: 1222–1229.
Kim HL, Kim SU, Seo YR . A novel role for Gadd45alpha in base excision repair: modulation of APE1 activity by the direct interaction of Gadd45alpha with PCNA. Biochem Biophys Res Commun 2013; 434: 185–190.
Kutuzov MM, Ilina ES, Sukhanova MV, Pyshnaya IA, Pyshnyi DV, Lavrik OI et al. Interaction of poly(ADP-ribose) polymerase 1 with apurinic/apyrimidinic sites within clustered DNA damage. Biochemistry (Moscow) 2011; 76: 147–156.
Azam S, Jouvet N, Jilani A, Vongsamphanh R, Yang X, Yang S et al. Human glyceraldehyde-3-phosphate dehydrogenase plays a direct role in reactivating oxidized forms of the DNA repair enzyme APE1. J Biol Chem 2008; 283: 30632–30641.
Zhang ZM, Yang XQ, Wang D, Wang G, Yang ZZ, Qing Y et al. Nm23-H1 protein binds to APE1 at AP sites and stimulates AP endonuclease activity following ionizing radiation of the human lung cancer A549 cells. Cell Biochem Biophy 2011; 61: 561–572.
Yamamori T, DeRicco J, Naqvi A, Hoffman TA, Mattagajasingh I, Kasuno K et al. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res 2010; 38: 832–845.
Zhao J, Gao F, Zhang Y, Wei K, Liu Y, Deng X . Bcl2 inhibits abasic site repair by down-regulating APE1 endonuclease activity. J Biol Chem 2008; 283: 9925–9932.
Acknowledgements
This research was supported by the Alzheimer’s Association, USA under grant NIRG-11-203527 to AKM. ST acknowledges financial support from the Indian Council for Medical Research (ICMR), New Delhi, India for providing a Junior Research Fellowship (JRF). BS, RPC and NG acknowledge financial support from CUPB for providing an Institutional Fellowship. We thank Dr Alpna Saini of the Center for Comparative Literature, School of Languages, Literature and Culture, Central University of Punjab, Bathinda, India for proofreading the manuscript for typographic and linguistic errors. As a result of the limited focus of the article, many appropriate references could not be included, for which the authors apologize. AKM specially thanks Drs J Regino Perez-Polo and Sankar Mitra at the Department of Biochemistry and Molecular Biology, University of Texas Medical Branch, Galveston, TX, USA and their laboratory members for professional development over the years. The CUPB publication number provided for this review is P-102.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Thakur, S., Sarkar, B., Cholia, R. et al. APE1/Ref-1 as an emerging therapeutic target for various human diseases: phytochemical modulation of its functions. Exp Mol Med 46, e106 (2014). https://doi.org/10.1038/emm.2014.42
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2014.42
