
Abstract
Autophagy is a dynamic cellular pathway involved in the turnover of proteins, protein complexes, and organelles through lysosomal degradation. The integrity of postmitotic neurons is heavily dependent on high basal autophagy compared to non-neuronal cells as misfolded proteins and damaged organelles cannot be diluted through cell division. Moreover, neurons contain the specialized structures for intercellular communication, such as axons, dendrites and synapses, which require the reciprocal transport of proteins, organelles and autophagosomes over significant distances from the soma. Defects in autophagy affect the intercellular communication and subsequently, contributing to neurodegeneration. The presence of abnormal autophagic activity is frequently observed in selective neuronal populations afflicted in common neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, Huntington's disease and amyotrophic lateral sclerosis. These observations have provoked controversy regarding whether the increase in autophagosomes observed in the degenerating neurons play a protective role or instead contribute to pathogenic neuronal cell death. It is still unknown what factors may determine whether active autophagy is beneficial or pathogenic during neurodegeneration. In this review, we consider both the normal and pathophysiological roles of neuronal autophagy and its potential therapeutic implications for common neurodegenerative diseases.
Similar content being viewed by others

Introduction
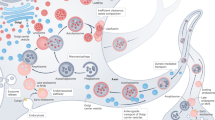
Neurons have highly specialized structures for intercellular communication, which typically include the axon, dendrites and synapses (Figure 1). In particular, the axon transfers proteins and organelles over significant distances in the nervous system. Almost all the materials contained in the axon originate in the soma and are localized to the axon via axoplasmic transport, in which the constant quality control of proteins and mitochondria is critical for proper neuronal function. Synapses represent regions of high energy demand and protein turnover; they contain abundant mitochondria and polyribosomes which make them more susceptible to the consequences of dysfunctional autophagy. In addition, the fact that neurons are postmitotic and do not replicate in general might predispose them to the accumulation of toxic proteins and damaged organelles inside neurons that could become diluted through cell division in replicating cells.

The basic and pathogenic role of neuronal autophagy. Neurons have highly specialized structures for intercellular communication, which typically include the soma, axon, dendrites and synapses. In the soma, the central region of the neuron containing the nucleus, basal levels of autophagy, including macroautophagy, mitophagy and chaperone-mediated autophagy (CMA), occur to maintain normal cellular homeostasis. The axon, a specialized structure to conduct nerve impulses, transfers proteins and organelles over significant distances by axoplasmic transport. Axotomy and excitotoxic insult trigger the accumulation of autophagosomes in dystrophic axonal swellings. Autophagic vaculoes (AVs) have also been observed in dysfunctional axons in alzheimer's disease (AD), parkinson's disease (PD) and huntington's disease (HD). Synapse, the region of contact where a neuron transferring information to another cell, represents a region of high energy demand and protein turnover; They contain abundant mitochondria and polyribosomes. Autophagy is known to play an important role in synapse development as autophagy is induced in response to many developmental and environmental cues. Thus, autophagy may play an important role in the synapse growth and plasticity required for learning and memory.
The critical importance of tightly controlled proteins and organelles in the neurons suggests that neuronal autophagy may be regulated distinctively from that of non-neuronal cells. In accordance with this notion, a study using GFP-LC3 transgenic mouse model demonstrated that autophagy is distinctly regulated in neurons (Mizushima et al., 2008). Other work has revealed significant differences in basal autophagy between neuronal and non-neuronal cells (Mitra et al., 2009). Recent loss-of function studies demonstrate a critical role for basal autophagy in the removal of damaged proteins and organelles in specific neurons of the brain (Hara et al., 2006; Komatsu et al., 2006).
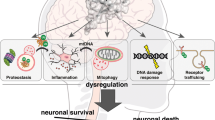
Intracellular protein aggregates and dysfunctional organelles are common features of neurodegenerative diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD) and amyotrophic lateral sclerosis (ALS). Subsequently, in the course of these diseases, toxic protein aggregates and damaged organelles accumulate within specific types of neurons and lead to neuronal dysfunction and ultimate neuronal death (Figure 2). Therefore, the precise and localized activation of autophagy in soma, axons or synapses may represent a beneficial therapeutic intervention for neurodegenerative diseases, even though potential pathogenic contribution of activated autophagy must be carefully monitored.

Disease causing proteins affecting various types of autophagy in common neurodegenerative diseases. Familial forms of the common neurodegenerative diseases, such as AD, PD, HD and ALS, are caused by gene mutations, which affect different types and steps of autophagy. AD is characterized by the pathogenic accumulation of amyloid plaques consisting of β-amyloid (Aβ) peptides generated by proteolytic cleavage of amyloid precursor protein (APP). AVs are a major reservoir of intracellular Aβ in the brain. Mutant presenilin 1 (PS1) impairs autophagosome clearance, resulting in enhanced Aβ accumulation in AD. PD is characterized by the presence of Lewy bodies, the intracytoplasmic inclusions containing α-synuclein, which is degraded by macroautophagy and CMA. The inhibition of CMA leads to an accumulation of soluble high molecular weight and detergent-insoluble species of α-synuclein and the inhibition of macroautophagy also leads to the accumulation of wild type α-synuclein. Mitochondrial dysfunction has been implicated in the pathogenesis of PD. PINK1 and parkin mediate the removal of mitochondria by mitophagy, of which mutations result in the accumulation of impaired mitochondria. HD is characterized by the presence of inclusion bodies composed of the N-terminal fragment of mutant Htt, which is associated with the primary defect in the ability of autophagic vacuoles to recognize cytosolic cargo. Mutations in the SOD1gene underlie 20% of familial ALS cases. Agents promoting autophagy, such as lithium and rapamycin, help autophagic clearance of mutant SOD1.
Characteristics of neuronal autophagy in the synapse and axon
Autophagy in synapse development
Neuronal autophagy plays an important role in synapse development. A recent study demonstrates that decreasing or increasing autophagy results in corresponding effects on synapse size (Shen and Ganetzky, 2009). They found that neuronal autophagy positively regulates synaptic development in the Drosophila neuromuscular junction. Overexpression of Atg1, a serine/threonine kinase and key regulator of autophagy, is sufficient to induce high levels of autophagy and subsequent enhancement of synaptic growth. In contrast, reducing autophagy activity through mutations in autophagy genes results in the reduction of synapse size. The ubiquitin-proteasome pathway system (UPS) is known to be an important negative regulator of synaptic development in the neuromuscular junction (Wan et al., 2000). Neuronal autophagy and UPS converge to regulate synapse development by regulating Hiw, an E3 ubiquitin ligase. Of interest, Atg1 is suggested to play a role in the regulation of synaptic structure via the downregulation of the MAP kinase ERK activity (Wairkar et al., 2009). In mice carrying an Atg1 mutation, synaptic density was decreased by excessive ERK activity, suggesting that activated ERK negatively regulates synapse formation. As autophagy is induced in response to many developmental and environmental cues, neuronal autophagy may play an important role in the synapse growth and plasticity required for learning and memory.
Axonal autophagy in injured neurons
The axon is a highly specialized structure dedicated to information transfer over sometimes lengthy distances in the nervous system. Autophagy has previously been suggested to be associated with axonal dystrophy following axotomy or excitotoxic stimuli. For instance, after axotomy, autophagosome-like vesicles accumulate in axon terminals (Dixon, 1967; Mattews and Raisman, 1972). This increased autophagy is accompanied by chromatolysis, a term that describes an area of cytoplasm that is devoid of organelles and filled with various types of vesicles including autophagosomes (Dixon, 1967). Similarly, in Purkinje cells of the Lurcher mouse, a model of excitotoxic neurodegeneration, excitotoxic insults trigger the accumulation of GFP-LC3-labeled autophagosomes in dystrophic axonal swellings, a hallmark of CNS axonopathy (Wang et al., 2006). Autophagosome-like vesicles have also been observed in dysfunctional axons in AD, PD and HD (Roizin et al., 1974; Anglade et al., 1997; Nixon et al., 2005).
Autophagy in Alzheimer's disease
AD is the most common form of senile dementia. AD is characterized by the pathogenic accumulation of amyloid plaques consisting of β-amyloid (Aβ) peptides, which are generated by the sequential cleavage of amyloid precursor protein (APP) via the β-secretase and γ-secretase, and by the intracellular accumulation of neurofibrillary tangles containing hyperphosphorylated tau (Vassar et al., 1999). Electron microscopy studies on AD brain provided the first evidence that autophagy is extensively involved in the neurodegenerative process (Nixon et al., 2005). Autophagic vacuoles (AVs) were abundant in AD brains, particularly, within dystrophic neuritis. Autophagy was also evident in the perikarya of affected neurons, especially in those with neurofibrillary tangles. The significant accumulations of immature AV forms in dystrophic neurites suggest that the transport of AVs and their maturation to lysosomes, may be impaired in AD. Subsequently, immunolabeling studies identified AVs in the brain to be a major reservoir of intracellular Aβ (Yu et al., 2005). Purified AVs contain both full-length APP and β-secretase cleaved APP and are highly enriched in the γ-secretase complex subunits presenilin-1 (PS1) and nicastrin. In addition, the mammalian target of rapamycin (mTOR) kinase-mediated modulation of autophagy in neuronal cells elicits parallel changes in AV proliferation and Aβ production. These data suggest a direct link between β-amyloidogenic and cell survival pathways via activated autophagy in AD.
Autophagosomes and endosomes within neurons actively form in synapses and along neuritic processes; however, their efficient clearance requires their retrograde transport towards the soma, which contains the highest concentration of lysosomes. In AD the maturation of autophagolysosomes and their retrograde transport are impaired, which results in a massive accumulation of AVs within degenerating neurites. This combination of increased autophagy induction and defective clearance of Aβ-generating AVs results in Aβ accumulation (Nixon, 2007; Boland et al., 2008). Furthermore, defective lysosomal proteolysis may underlie further pathogenic protein accumulations and neuronal cell death in AD. The AD-related protein PS1 is required for autophagy and in neurons from mice hypomorphic for PS1, autophagosome clearance is prevented as a result of the selective impairment of autolysosome acidification and cathepsin activation, which are caused by the failure of PS-1-dependent targeting of v-ATPase subunit to lysosomes (Lee et al., 2010). N-glycosylation of the v-ATPase subunit is essential for its efficient ER-to-lysosome delivery, which requires the selective binding of PS1 to its unglycosylated subunit. Similarly, PS1 mutations responsible for early-onset AD produce a defective lysosomal/autophagy phenotype in fibroblasts of AD patients. These phenomena could be explained by the fact that PS1 is essential for the targeting of v-ATPase subunit to lysosomes, lysosome acidification, and proteolysis during autophagy (Cataldo et al., 2004; Lee et al., 2010). These findings show that the impairment of autophagosome maturation could lead to the increased accumulation of Aβ in AD.
Beclin1, also known as Atg6, is a proautophagic protein. The protein Beclin1 expression decreases in aged human brain, leading to a reduction of autophagy with aging. Beclin 1 has been shown to be decreased in the AD brains (Pickford et al., 2008). Decreased neuronal autophagy and subsequent neurodegeneration is observed in mice carrying a heterozygous deletion of beclin 1 (Pickford et al., 2008). In the APP transgenic mouse model of AD, the reduced expression of beclin1 results in an increase of intraneuronal Aβ accumulation, extracellular Aβ deposition and neurodegeneration, which was ameliorated by the lentiviral expression of beclin 1 (Pickford et al., 2008). Moreover, beclin1 deficiency has been shown to disrupt cellular autophagy and autophagosomal-lysosomal degradation and to alter APP metabolism (Jaeger et al., 2010). These studies suggest that reduced autophagy via beclin1 deficiency might reduce Aβ clearance; therefore, increasing beclin 1 levels may have a therapeutic potential in AD.
The mTOR pathway plays an essential role in neuronal functions by controlling protein homeostasis. mTOR signaling has been shown to regulate various forms of learning and memory. In an animal model of AD, the pharmacological inhibition of mTOR signaling with rapamycin rescues cognitive deficits and ameliorates Aβ and tau pathology by increasing autophagy (Caccamo et al., 2010). Similarly, the long-term inhibition of mTOR by rapamycin prevents AD-like cognitive deficits and lowered levels of Aβ in the PDAPP transgenic mouse model (Spilman et al., 2010). These results provide a molecular basis for the efficacy of mTOR inhibition in ameliorating cognitive impairment in AD models by demonstrating that the resultant increased autophagy results in the reduction in Aβ and the improvement in cognitive function. These observations collectively suggest that autophagy plays a critical role in the pathogenesis of AD and that the inhibition of mTOR signaling and/or the activation of beclin 1 signaling may reduce Aβ and tau pathology, subsequently improving learning and memory in AD patients.
Autophagy in Parkinson's disease
PD is one of the most common neurodegenerative disorders. PD is caused by the selective cell death of dopamine neurons in the substantia nigra (SN) and is characterized by the presence of Lewy bodies, which are the intracytoplasmic inclusions containing α-synuclein and ubiquitin. Early ultrastructural examinations in PD patients revealed characteristics of apoptosis and autophagy in the degenerating melanized neurons of the SN (Anglade et al., 1997). Subsequently, the pathogenic role of autophagy in PD was demonstrated by the finding that α-synuclein, a major constituent of Lewy body, is degraded by macroautophagy and chaperone-mediated autophagy (CMA) (Webb et al., 2003; Cuevo et al., 2004; Vogiatzi et al., 2008). The inhibition of CMA leads to an accumulation of soluble high molecular weight and detergent-insoluble species of α-synuclein and the inhibition of macroautophagy also leads to the accumulation of wild type α-synuclein, suggesting that CMA and macroautophagy are important pathways for wild type α-synuclein degradation in neurons (Vogiatzi et al., 2008). In addition, beclin 1 plays an important role in the intracellular degradation of α-synuclein either directly or indirectly through the autophagic pathway. When a lentivirus expressing beclin 1 was delivered to the brain of an α-synuclein transgenic mouse, the synaptic and dendritic pathology was ameliorated and the accumulation of α-synuclein in the limbic system was reduced (Spencer et al., 2009). These results were accompanied by enhanced lysosomal activation and a reduction in deficits in the autophagy pathway.
The familial PD-associated molecules, such as the wild type, A53T and A30P mutants of α-synuclein, DJ-1, parkin, PINK1 and LRRK2 are known to be involved in the autophagic pathway. For instance, overexpression of wild type α-synuclein impairs macroautophagy both in vitro, in mammalian cells and in vivo, in transgenic mice because multiplication of the α-synuclein gene locus is known to increase α-synuclein expression and cause familial PD (Winslow et al., 2010). α-Synuclein overexpression causes the mislocalization of the autophagy protein, Atg9, and decreased formation of omegasome. Both α-synuclein, and Atg9 regulate formation of the omegasomes, marking autophagosome precursors. Of interest, A53T and A30P mutants inhibit CMA via a higher binding affinity for the lysosomal-associated membrane protein 2A (LAMP2A) compared with wild type α-synuclein (Cuevo et al., 2004; Xilouri et al., 2009). Overexpression of both mutant and wild type α-synuclein is accompanied by the induction of compensatory macroautophagy. The mutant α-synuclein is degraded by macroautophagy instead of CMA. Therefore, impaired CMA might trigger neuronal death and the preservation of proper CMA activity could be a potential target for PD therapeutics. The functional deficiency of DJ-1, a recessive familial PD gene, also leads to altered autophagy (i.e. increased autophagic flux) in murine and human cells (Irrcher et al., 2010). In addition, mutations in the leucine-rich repeat kinase 2 (LRRK2) have been implicated in both autosomal dominant and some cases of sporadic PD. Cells transfected with G2019S LRRK2 mutant exhibit striking increases of autophagic vacuoles in both neuritic and somatic compartments (Plowey et al., 2008). Knockdown of LC3 or Atg7, two essential components of the autophagic machinery, reversed the effects of G2019S LRRK2 expression on neuronal process length, whereas rapamycin potentiated these effects, suggesting that the pathogenic mutation of LRRK2 plays active role for autophagy in neurite remodeling. Mutations in the PTEN-induced putative kinase 1 (PINK1) gene also cause autosomal recessive PD. The full-length PINK1 interacts with Beclin1 and the overexpression of PINK1 significantly enhances both basal and starvation-induced autophagy, which can be reduced by beclin1 gene knockdown. Of interest, the W437X mutant of PINK1 demonstrates an impaired interaction with Beclin1 and lacks the ability to induce autophagy, whereas the G309D mutant of PINK1, which has defective kinase activity, does bind with Beclin1 (Michiorri et al., 2010).
Although neuronal autophagy appears primarily to be a protective process in the nervous system, it can also play a paradoxical role in neuronal death. With respect to the role of autophagy in neuronal death, several studies employing PD toxins, a mutant familial PD gene and postmortem PD brains have demonstrated an important role for autophagy in promoting the death of dopamine neurons. For example, autophagic cell death has been observed in nigral dopamine neurons of PD patients (Anglade et al., 1997). MPP+ or dopamine toxicity-induced oxidative stress, increases the number of AVs, autophagy and cell death, all of which differs from what is observed in starvation-induced autophagy (Gomez-Santos et al., 2003; Zhu et al., 2007). Oxidative stress, a major pathogenic mechanism of PD, has been shown to increase autophagic cell death in dopaminergic neurons by suppressing the expression of Oxi-α gene, which encodes a novel mTOR activator (Choi et al., 2010). The Oxi-α gene knockdown resulted in not only the repression of mTOR kinase activity, but also enhances neuronal susceptibility to oxidative stress. In agreement with these observations, treatment with rapamycin, an mTOR inhibitor and autophagy inducer, potentiates cell death induced by oxidative stress. In addition, overexpression of the A53T mutant of human α-synuclein induces an accumulation of AVs and autophagic cell death (Stefanis et al., 2001; Kirik et al., 2002; Xilouri et al., 2009; Yang et al., 2009). These studies suggest that pathogenic autophagy associated with neuronal death does occur and may be distinct from basal neuronal autophagy. The contribution of autophagy and autophagic cell death to degeneration of dopamine neurons may vary depending on the initial cause and specific cellular context. A better understanding of autophagic stress (Chu, 2006) and further identification of autophagic cell death mechanisms may lead to therapeutics that help restore homeostasis to dopamine neurons in PD.
Damaged mitochondria accumulate with age and mitochondrial dysfunction has been implicated in the pathogenesis of PD. Previously, recessive mutations in parkin, an ubiquitin E3 ligase, have been identified as a cause of early onset, familial PD,. However, the precise role of parkin in mitochondria is unknown. It was recently shown that parkin is selectively recruited to damaged mitochondria, which have low membrane potential in mammalian cells. Following recruitment, parkin mediates the removal of mitochondria by autophagy and the selective elimination of impaired mitochondria (Narendra et al., 2008). This discovery revealed a link between the mitochondrial quality control and proteins mutated in familial PD, and further implicates a failure to eliminate dysfunctional mitochondria in the pathogenesis of PD.
The link between parkin and mitophagy has brought about great interest in the molecular mechanism of mitophagy as well as the pathogenic role of mitophagy in PD (Figure 3). Of interest, PINK1, a mitochondrial serine/threonine kinase, also implicated in familial PD, is normally maintained at low levels on mitochondria by voltage-dependent proteolysis (Narendra et al., 2010), mediated by the mitochondrial presenilin-associated rhomboid-like protein (PARL) present in the inner mitochondrial membrane (Jin et al., 2010). In the absence of PARL, the constitutive degradation of PINK1 is inhibited and a 60-kD form of PINK1 is stabilized inside mitochondria. When the mitochondrial membrane potential is dissipated, PINK1 accumulates as a 63-kD full-length form in the outer mitochondrial membrane. Thus, damage to mitochondria facilitates the rapid accumulation of PINK1 and the subsequent PINK1 accumulation in the mitochondria recruits parkin to induce mitophagy (Narendra et al., 2010; Geisler et al., 2010). At this stage, the autophagic adaptor protein p62/SQSTM1 is recruited to mitochondria, which may be important for the clearance of mitochondria (Figure 3). In addition, the voltage-dependent anion channel 1 (VDAC1) is a target for parkin-mediated poly-ubiquitylation of Lys 27 and mitophagy (Geisler et al., 2010). Thus, pathogenic parkin mutations, together with PINK1 mutations, could lead to the disruption of mitochondrial recruitment of parkin, ubiquitination of mitochondrial substrates, formation of AVs and the final clearance of damaged mitochondria via mitophagy. Consistent with this notion, a detailed analysis of PINK1 and parkin knockout models revealed an accumulation of damaged mitochondria in various tissues including dopamine neurons (Greene et al., 2003; Gautier et al., 2008; Shim et al., 2011). These observations suggest that neuronal autophagy is essential for the turnover of damaged mitochondria in PD and that the failure to induce mitophagy may underlie the selective dopaminergic neuronal loss observed in PD. Thus, stimulating the proper induction of mitophagy in dopaminergic neurons may serve as a therapeutic target to slow disease progression in PD.

PINK1 and parkin-mediated mitophagy. In the current model of the PINK1/parkin-mediated mitophagy, PINK1, implicated in familial PD, is normally maintained at low levels on mitochondria by voltage-dependent proteolysis, which is mediated by the mitochondrial presenilin-associated rhomboid-like protein (PARL) present in the inner mitochondrial membrane. In the absence of PARL, the constitutive degradation of PINK1 is inhibited and a 60-kD form of PINK1 is stabilized inside mitochondria. When the mitochondrial membrane potential is dissipated, PINK1 accumulates as a 63-kD full-length form in the outer mitochondrial membrane. Thus, accumulation of mitochondrial damage by gene mutations or carbonyl cyanide m-chloro phenyl hydrazone (CCCP), a mitochondrial uncoupling agent, facilitates the rapid accumulation of PINK1 and the subsequent PINK1 accumulation in the mitochondria recruits parkin to ubiquitinate VDAC (and/or mitofusin) on the mitochondrial outer membrane, which becomes a target for mitophagy. The ubiquitin-binding adaptor p62 (also known as sequestosome 1) recruits ubiquitinated cargo into autophagosomes by binding to LC3.
Autophagy in Huntington's disease
HD, an autosomal dominant movement disorder, is caused by the expansion (>35 repeats) of CAG trinucleotide repeats in the huntingtin (htt) gene, which encodes an expanded polyglutamine (poly(Q)) tract in the N-terminus of the Htt protein. The toxicity of the mutant Htt protein is believed to result from the intraneuronal aggregates of the N-terminal fragments, the typical pathological hallmark of HD. The characteristic features of altered autophagy were first observed in postmortem brains of HD patients (Tellez-Nagel et al., 1974). In the models of HD, when mutant Htt was exogenously expressed in striatal neurons, mutant Htt-labeled vacuoles displayed the ultrastructural features of autophagosomes. In addition, neurons with excess cytoplasmic Htt exhibited an increased tubulation of endosomal membranes (Kegel et al., 2000). Thus, the accumulation of mutant Htt might activate the endosomal-lysosomal system and contribute to an autophagic process of cell death. The involvement of autophagy in HD was further demonstrated by the sequestration of mTOR in polyglutamine aggregates in cell models, transgenic mice and human brains. The sequestration of mTOR impairs its kinase activity and induces autophagy. This autophagy protects against polyglutamine toxicity, as rapamycin attenuates Htt accumulation and cell death in cell models of HD and the inhibition of autophagy has the opposite effects (Ravikumar et al., 2004). Subsequently, a number of studies probed the therapeutic potential of autophagy induction in various models of HD (Ravikumar et al., 2003, 2004; Sakar et al., 2007).
The precise mechanism that underlies autophagic dysfunction in HD is poorly understood. The inefficient engulfment of cytosolic components by autophagosomes has been proposed to be responsible for their slower turnover and the accumulation of Htt in HD cells. Using cellular and mouse models of HD and cells from HD patients, a primary defect in the ability of autophagic vacuoles to recognize cytosolic cargo, was identified in HD cells. Autophagic vacuoles formed at normal rates and were adequately eliminated by lysosomes, however, they failed to efficiently trap cytosolic cargo in their lumen (Matinez-Vincente et al., 2010). Another mechanism underlying the intracellular accumulation of mutant Htt is that the accumulated mutant Htt recruits beclin 1 and impairs the beclin 1-mediated long lived protein turnover. Thus, the sequestration of beclin 1 in the vulnerable neuronal population of HD patients might further reduce beclin 1 function and autophagic degradation of mutant Htt (Shibata et al., 2006). As beclin 1 expression decreases in an age-dependent fashion in the human brains and the heterozygous deletion of beclin 1 gene is insufficient for the regulation of autophagosomes, it has been proposed that the age-dependent decrease of beclin 1 expression may lead to a reduction of autophagic activity, which in turn promotes the accumulation of mutant Htt and the progression of the disease (Shibata et al., 2006).
One homeostatic function of the wild type Htt protein has been proposed to be to act as an endoplasmic reticulum (ER) sentinel, regulating autophagy in response to ER stress (Atwal et al., 2008). As a result, cells expressing mutant Htt may have perturbed ER function and have increased autophagic vesicles. Moreover, a number of findings suggest that the Htt protein exhibits structural and functional similarities to the central autophagy regulator, mTOR. For instance, Htt and mTOR are both scaffolding proteins and contain similar leucine-rich repeats, termed HEAT domains (Andrade and Bork, 1995). Both Htt and mTOR use amino-terminal HEAT repeats to specifically recognize membranous structures. In this way, mTOR reversibly associates with ER and Golgi membranes (Drenan et al., 2004). Similarly, Htt reversibly associate with ER and autophagic vesicles (Omi et al., 2005). As a result, cells expressing mutant Htt may have a perturbed ER and an increase in AVs (Atwal et al., 2008). Therefore, these observations imply a potential role of Htt in the regulation of autophagy.
The initial increase in AVs and autophagy observed in HD models may represent an attempt to remove mutant Htt protein and over time the autophagy machinery becomes dysfunctional, leading to neurodegeneration. Thus, the therapeutic induction and recovery of autophagy might may enhance the clearance of mutant Htt protein and reduce its toxic effect in HD neurons.
Autophagy in amyotrophic lateral sclerosis
ALS is a progressive neurodegenerative disorder that selectively affects motor neurons. Approximately 10% of ALS cases are familial ALS (fALS) and, mutations in the Cu, Zn-superoxide dismutase (SOD1) gene underlie 20% of fALS cases, suggesting its crucial role in the pathogenesis of fALS. Although various pathogenic mechanisms have been proposed for ALS, a possible role for autophagy was observed in transgenic mouse models of fALS. For instance, in the transgenic SOD1G93A mouse, increased autophagy was observed during the presymptomatic stage (<90 days of age). Surprisingly, any significant changes in autophagy in the soma of motor neurons were not observed until the terminal stage in these mice (140 days of age) (Li et al., 2008). Another study also demonstrated that autophagosome formation was increased at the symptomatic stage and the percentage of motor neurons that contained phosphorylated mTOR/Ser2448 was decreased in the SOD1G93A transgenic mice (Morimoto et al., 2007), which suggests the possibility that increased and/or impaired autophagy underlies pathological phenomena in this model. Much uncertainty still remains on the pathogenic mechanisms underlying motor neuron degeneration in ALS; Riluzole is the only available drug for the treatment of ALS and has marginal effects on disease survival. A number of putative therapeutic agents have recently been investigated for their potential role in neuroprotection (Siciliano et al., 2010). Among these, agents promoting autophagy, such as lithium and rapamycin, have been suggested to be effective for the survival of motor neurons in ALS by autophagic clearance of SOD1. Additional studies to address the precise pathogenic mechanism of autophagy in motor neuron death are essential for the identification of therapeutic targets in ALS.
Conclusion
Mounting evidence has firmly established the importance of neuronal autophagy in both the normal functioning and patho-physiological conditions, of the brain. Neuronal integrity is more sensitive to alterations in basal autophagy than that of non-neuronal cells due to their postmitotic nature and the specialized structures necessary for intercellular communications. Impairments in the quality control of proteins and organelles in the neuronal soma, axon and synapses likely disrupt normal neuronal function due to the accumulation of toxic protein aggregates and damaged organelles, ultimately leading to neurodegenerative conditions observed in diseases, such as AD, PD, HD and ALS. However, the cellular factors underlying homeostatic vs. pathogenic activation of autophagy have not been identified, nor is it understood how dysfunctional autophagy leads to neuronal death. Under certain pathophysiological conditions autophagy may represent a cell death mechanism via excessive removal of key survival factors or cellular organelles. More importantly, protein aggregates and damaged organelles picked up by autophagosomes in axon and synapses must be transported over distances to lysosomes in the soma, which implies the involvement of complex processes in neuronal autophagy. Therefore, an improved understanding of neuronal autophagy will provide novel insights into the pathogenic mechanisms of dysfunctional autophagy that underlie common neurodegeneratives diseases and, ultimately, help develop therapeutic interventions for autophagic dysregulation.
Abbreviations
- AD:
-
Alzheimer's disease
- ALS:
-
amyotrophic lateral sclerosis
- HD:
-
Huntington's disease
- mTOR:
-
mammalian target of rapamycin
- PD:
-
Parkinson's disease
References
Andrade MA, Bork P . HEAT repeats in the Huntington's disease protein . Nat Genet 1995 ; 11 : 115 - 116
Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J . Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease . Histol Histopathol 1997 ; 12 : 25 - 31
Atwal RS, Truant R . A stress sensitive ER membrane-association domain in Huntingtin protein defines a potential role for Huntingtin in the regulation of autophagy . Autophagy 2008 ; 4 : 91 - 93
Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH . Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease . J Neurosci 2008 ; 28 : 6926 - 6937
Caccamo A, Majumder S, Richardson A, Strong R, Oddo S . Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments . J Biol Chem 2010 ; 285 : 13107 - 13120
Cataldo AM, Peterhoff CM, Schmidt SD, Terio NB, Duff K, Beard M . Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology . J Neuropathol Exp Neurol 2004 ; 63 : 821 - 830
Choi KC, Kim SH, Ha JY, Kim ST, Son JH . A novel mTOR activating protein protects dopamine neurons against oxidative stress by repressing autophagy related cell death . J Neurochem 2010 ; 112 : 366 - 376
Chu CT . Autophagic stress in neuronal injury and disease . J Neuropathol Exp Neurol 2006 ; 65 : 423 - 432
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D . Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy . Science 2004 ; 305 : 1292 - 1295
Dixon JS . "Phagocytic" lysosomes in chromatolytic neurones . Nature 1967 ; 215 : 657 - 658
Drenan RM, Liu X, Bertram PG, Zheng XF . FKBP12-rapamycin-associated protein or mammalian target of rapamycin (FRAP/mTOR) localization in the endoplasmic reticulum and the Golgi apparatus . J Biol Chem 2004 ; 279 : 772 - 778
Gautier CA, Kitada T, Shen J . Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress . Proc Natl Acad Sci USA 2008 ; 105 : 11364 - 11369
Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ . PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1 . Nat Cell Biol 2010 ; 12 : 119 - 131
Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S . Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells . J Neurosci Res 2003 ; 73 : 341 - 350
Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ . Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants . Proc Natl Acad Sci USA 2003 ; 100 : 4078 - 4083
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R . Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice . Nature 2006 ; 441 : 885 - 889
Irrcher I, Aleyasin H, Seifert EL, Hewitt SJ, Chhabra S, Phillips M . Loss of the Parkinson's disease-linked gene DJ-1 perturbs mitochondrial dynamics . Hum Mol Genet 2010 ; 19 : 3734 - 3746
Jaeger PA, Pickford F, Sun CH, Lucin KM, Masliah E, Wyss-Coray T . Regulation of amyloid precursor protein processing by the Beclin 1 complex . PLoS One 2010 ; 5 : e11102 -
Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ . Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL . J Cell Biol 2010 ; 191 : 933 - 942
Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N . Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy . J Neurosci 2000 ; 20 : 7268 - 7278
Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N . Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system . J Neurosci 2002 ; 22 : 2780 - 2791
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I . Loss of autophagy in the central nervous system causes neurodegeneration in mice . Nature 2006 ; 441 : 880 - 884
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM . Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations . Cell 2010 ; 141 : 1146 - 1158
Li L, Zhang X, Le W . Altered macroautophagy in the spinal cord of SOD1 mutant mice . Autophagy 2008 ; 4 : 290 - 293
Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S . Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease . Nat Neurosci 2010 ; 13 : 567 - 576
Matthews MR, Raisman G . A light and electron microscopic study of the cellular response to axonal injury in the superior cervical ganglion of the rat . Proc R Soc Lond B Biol Sci 1972 ; 181 : 43 - 79
Michiorri S, Gelmetti V, Giarda E, Lombardi F, Romano F, Marongiu R . The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy . Cell Death Differ 2010 ; 17 : 962 - 974
Mitra S, Tsvetkov AS, Finkbeiner S . Protein turnover and inclusion body formation . Autophagy 2009 ; 5 : 1037 - 1038
Mizushima N, Levine B, Cuervo AM, Klionsky DJ . Autophagy fights disease through cellular self-digestion . Nature 2008 ; 451 : 1069 - 1075
Morimoto N, Nagai M, Ohta Y, Miyazaki K, Kurata T, Morimoto M . Increased autophagy in transgenic mice with a G93A mutant SOD1 gene . Brain Res 2007 ; 1167 : 112 - 117
Narendra D, Tanaka A, Suen DF, Youle RJ . Parkin is recruited selectively to impaired mitochondria and promotes their autophagy . J Cell Biol 2008 ; 183 : 795 - 803
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J . PINK1 is selectively stabilized on impaired mitochondria to activate Parkin . PLoS Biol 2010 ; 8 : e1000298 -
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A . Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study . J Neuropathol Exp Neurol 2005 ; 64 : 113 - 122
Nixon RA . Autophagy, amyloidogenesis and Alzheimer disease . J Cell Sci 2007 ; 120 : 4081 - 4091
Omi K, Hachiya NS, Tokunaga K, Kaneko K . siRNA-mediated inhibition of endogenous Huntington disease gene expression induces an aberrant configuration of the ER network in vitro . Biochem Biophys Res Commun 2005 ; 338 : 1229 - 1235
Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA . The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice . J Clin Invest 2008 ; 118 : 2190 - 2199
Plowey ED, Cherra SJ, Liu YJ, Chu CT . Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells . J Neurochem 2008 ; 105 : 1048 - 1056
Ravikumar B, Stewart A, Kita H, Kato K, Duden R, Rubinsztein DC . Raised intracellular glucose concentrations reduce aggregation and cell death caused by mutant huntingtin exon 1 by decreasing mTOR phosphorylation and inducing autophagy . Hum Mol Genet 2003 ; 12 : 985 - 994
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG . Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease . Nat Genet 2004 ; 36 : 585 - 595
Roizin L, Stellar S, Willson N, Whittier J, Liu JC . Electron microscope and enzyme studies in cerebral biopsies of Huntington's chorea . Trans Am Neurol Assoc 1974 ; 99 : 240 - 243
Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL . Small molecules enhance autophagy and reduce toxicity in Huntington's disease models . Nat Chem Biol 2007 ; 3 : 331 - 338
Shen W, Ganetzky B . Autophagy promotes synapse development in Drosophila . J Cell Biol 2009 ; 187 : 71 - 79
Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T . Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1 . J Biol Chem 2006 ; 281 : 14474 - 14485
Shim JH, Yoon SH, Kim KH, Han JY, Ha JY, Hyun DH . The antioxidant Trolox helps recovery from the familial Parkinson's disease-specific mitochondrial deficits caused by PINK1- and DJ-1-deficiency in dopaminergic neuronal cells . Mitochondrion 2011 ; 11 : 707 - 715
Siciliano G, Carlesi C, Pasquali L, Piazza S, Pietracupa S, Fornai F . Clinical trials for neuroprotection in ALS . CNS Neurol Disord Drug Targets 2010 ; 9 : 305 - 313
Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R . Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases . J Neurosci 2009 ; 29 : 13578 - 13588
Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D . Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease . PLoS One 2010 ; 5 : e9979 -
Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA . Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death . J Neurosci 2001 ; 21 : 9549 - 9560
Steiner H, Capell A, Leimer U, Haass C . Genes and mechanisms involved in beta-amyloid generation and Alzheimer's disease . Eur Arch Psychiatry Clin Neurosci 1999 ; 249 : 266 - 270
Tellez-Nagel I, Johnson AB, Terry RD . Studies on brain biopsies of patients with Huntington's chorea . J Neuropathol Exp Neurol 1974 ; 33 : 308 - 332
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P . Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE . Science 1999 ; 286 : 735 - 741
Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L . Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells . J Biol Chem 2008 ; 283 : 23542 - 23556
Wairkar YP, Toda H, Mochizuki H, Furukubo-Tokunaga K, Tomoda T, Diantonio A . Unc-51 controls active zone density and protein composition by downregulating ERK signaling . J Neurosci 2009 ; 29 : 517 - 528
Wan HI, DiAntonio A, Fetter RD, Bergstrom K, Strauss R, Goodman CS . Highwire regulates synaptic growth in Drosophila . Neuron 2000 ; 26 : 313 - 329
Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP . Induction of autophagy in axonal dystrophy and degeneration . J Neurosci 2006 ; 26 : 8057 - 8068
Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC . Alpha-Synuclein is degraded by both autophagy and the proteasome . J Biol Chem 2003 ; 278 : 25009 - 25013
Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA . alpha-Synuclein impairs macroautophagy: implications for Parkinson's disease . J Cell Biol 2010 ; 190 : 1023 - 1037
Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L . Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy . PLoS One 2009 ; 4 : e5515 -
Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ . Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy . Science 2009 ; 323 : 124 - 127
Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH . Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer's disease . J Cell Biol 2005 ; 171 : 87 - 98
Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT . Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death . Am J Pathol 2007 ; 170 : 75 - 86
Acknowledgements
This research was supported in part by a grant (2010-0009233) from NRF and a grant (2011-0006244) from NCRC program, the Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Son, J., Shim, J., Kim, KH. et al. Neuronal autophagy and neurodegenerative diseases. Exp Mol Med 44, 89–98 (2012). https://doi.org/10.3858/emm.2012.44.2.031
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2012.44.2.031
Keywords
This article is cited by
-
Inhibition of miR-542-3p augments autophagy to promote diabetic corneal wound healing
Eye and Vision (2024)
-
Reduction of DHHC5-mediated beclin 1 S-palmitoylation underlies autophagy decline in aging
Nature Structural & Molecular Biology (2024)
-
Doxycycline inhibits dopaminergic neurodegeneration through upregulation of axonal and synaptic proteins
Naunyn-Schmiedeberg's Archives of Pharmacology (2023)
-
Expression patterns of prosaposin and its receptors, G protein–coupled receptor (GPR) 37 and GPR37L1 mRNAs, in the chick inner ear
Cell and Tissue Research (2023)
-
LYST deficiency impairs autophagic lysosome reformation in neurons and alters lysosome number and size
Cellular and Molecular Life Sciences (2023)
