
Abstract
Cardiomyocytes can resist ischemia/reperfusion (I/R) injury through ischemic postconditioning (IPoC) which is repetitive ischemia induced during the onset of reperfusion. Myocardial ischemic preconditioning up-regulated protein 2 (MIP2) is a member of the WD-40 family proteins, we previously showed that MIP2 was up-regulated during ischemic preconditioning (IPC). As IPC and IPoC engaged similar molecular mechanisms in cardioprotection, this study aimed to elucidate whether MIP2 was up-regulated during IPoC and contributed to IPoC-mediated protection against I/R injury. The experiment was conducted on two models, an in vivo open chest rat coronary artery occlusion model and an in vitro model with H9c2 myogenic cells. In both models, 3 groups were constituted and randomly designated as the sham, I/R and IPoC/hypoxia postconditioning (HPoC) groups. In the IPoC group, after 45 min of ischemia, hearts were allowed three cycles of reperfusion/ischemia phases (each of 30 s duration) followed by reperfusion. In the HPoC group, after 6 h of hypoxia, H9c2 cells were subjected to three cycles of 10 minute reoxygenation and 10 minute hypoxia followed by reoxygenation. IPoC significantly reduced the infarct size, plasma level of Lactate dehydrogenase and creatine kinase MB in rats. 12 h after the reperfusion, MIP2 mRNA levels in the IPoC group were 10 folds that of the sham group and 1.4 folds that of the I/R group. Increased expression of MIP2 mRNA and attenuation of apoptosis were similarly observed in the HPoC group in the in vitro model. These effects were blunted by transfection with MIP2 siRNA in the H9c2 cells. This study demonstrated that IPoC induced protection was associated with increased expression of MIP2. Both MIP2 overexpression and MIP2 suppression can influence the IPoC induced protection.
Similar content being viewed by others


Introduction
Reperfusion therapy is the gold standard in the treatment of acute ischemia and infarction, but restoration of blood flow to ischemic tissue results in the paradoxical phenomenon known as ischemia/reperfusion (I/R) injury. I/R injury could be altered by endogenous protective strategies applied at different time points (Yellon and Hausenloy, 2007), and two endogenous protections were investigated most: ischemic preconditioning (IPC) and ischemic postconditioning (IPoC). IPC is defined as the cardioprotection conferred by multiple and brief episodes of occlusion/reperfusion (intermittent hypoperfusion) against the development of irreversible damage following a subsequent, more severe ischemic episode (Murry et al., 1986). IPoC denotes a condition in which a decrease in the infarct size results from brief periods of ischemia alternating with brief periods of reperfusion applied immediately during reperfusion following an ischemic insult (Zhao et al., 2003; Vinten-Johansen, 2007; Hausenloy and Yellon, 2008). The time of IPC limits it application, unlike IPC, any cardiac revascularization procedure in various clinical scenarios (coronary angioplasty, on pump cardiac surgery, and cardiac transplantation) is a potential target for IPoC (Staat et al., 2005). It has been demonstrated that an IPoC-like phenomenon (hypoxia postconditioning, HPoC) can be elicited after hypoxia and reoxygenation in cell culture (Sun et al., 2005; Wang et al., 2006).
Both IPC and IPoC harness the innate cardioprotective mechanisms. The exact mechanism behind the protective effect of IPC and IPoC still remains largely speculative. Emerging studies suggest that a common cardioprotective pathway presents at the time of myocardial reperfusion. They both seem to engage similar molecular mechanisms such as G-protein-coupled receptors and protein kinase cascades that converge on the mitochondrial permeability transition pore (mPTP), although with subtle differences before and after the ischaemia (Hausenloy and Yellon, 2004; Hausenloy et al., 2005; Hausenloy and Yellon, 2006; Vinten-Johansen, 2007; Hausenloy and Yellon, 2008).
During myocardial IPC, many genes such as HSP40, HSP70, Ferritin subunit H, and Adprt are up-regulated, HSP40 and HSP70 help damaged proteins to repair and prevent denatured protein aggregation; Ferritin prepares the heart against future high levels of 'free' iron, thus reducing the degree of myocardial damage after prolonged ischemia; Adprt serves as a molecular switch between the transcription and repair of DNA to avoid expression of damaged genes. All of these up-regulated genes participate in the endogenous cardioprotection against myocardial I/R injury (Joazeiro and Hunter, 2000; Latchman, 2001; Berenshtein et al., 2002).
Recently, we found a novel gene myocardial ischemic preconditioning up-regulated protein 2 (MIP2) which was up-regulated during IPC in rat heart. Subsequently, we found MIP2 was a member of WD-40 family proteins (Yuan et al., 2003; Jiang et al., 2008). WD-40 family proteins are a group of eukaryotic proteins that have been highly conserved in evolution across different species from yeast, fly, mouse, rat to human. WD-40 family member contains 4 to 8 repeating sequences that are separated by approximately 40 amino acids. A WD-40 repeat consists of 2 signature sites: a poorly conserved site A containing glycine and histidine (GH) pairs, and a well-conserved site B containing tryptophan and aspartate (WD) pairs (van der Voorn and Ploegh, 1992; Neer et al., 1994). Members of WD-40 family proteins play a variety of roles in intracellular signaling, RNA processing and degradation, gene expression, vesicular traffic and fusion, cytoskeletal assembly, and the cell cycle (Smith et al., 1999; Yu et al., 2000; Cabrera-Vera et al., 2003). Our previous study indicated that MIP2 was expressed abundantly in the heart and skeleton muscles, and increased myocardial MIP2 expression was associated with IPC. MIP2 was up-regulated by oxidative stress, MIP2 overexpression down-regulated the transcriptional activity of AP-1 and suppresses H2O2-induced cell death in neural cells (Yuan et al., 2003; Zhao et al., 2009; Wei et al., 2010).
MIP2 was up-regulated during IPC, like other genes as mentioned above, MIP2 may provide protection against cardiomyocyte apoptosis. Many studies suggested that the mechanism behind the protective effect of IPC and IPoC engaged similar molecular mechanisms. Thus, we speculate that MIP2 might be up-regulated during IPoC and play an important role in the cardioprotection induced by IPoC.
Results
Myocardial I/R injury was significantly reduced by IPoC
Cardiac infarction due to I/R was evaluated by 2, 3, 5-triphenyl-tetrazolium chloride (TTC) staining. Representative sections (Figure 1A) demonstrated I/R induced infarction (the pale area) was attenuated in the IPoC group. Infarction was not detected in the sham group. Infarction was reduced to 16.1 ± 1.3% in the IPoC group compared with 31.7 ± 0.5% in the I/R group (P < 0.05). In addition, Plasma leakage of Lactate dehydrogenase (LDH) and creatine kinase MB (CK-MB) in the IPoC group was suppressed compared with that in the I/R group (P < 0.05). Both LDH and CK-MB activities was diminished at 24 h, with no obvious changes in LDH or CK-MB release in the sham group (Figures 1B and 1C).

IPoC protects I/R induced myocardial injury. (A) Pictures showed transverse sections of TTC stained heart from SD rats in the sham, I/R and IPoC groups. The infarct areas are pale and the viable tissues are red. Graphs showed the level of plasma CK-MB (B) and LDH (C) during different time courses of the experiment. #P < 0.05 vs sham group, *P < 0.05 vs I/R group. Values are means ± SEM.
MIP2 expression was enhanced by IPoC in the in vivo rat model
The level of MIP2 mRNA was assessed in the 3 groups between 1 and 24 h after the reperfusion. IPoC group induced significant MIP2 gene expression compared with the sham group and I/R group at any time point with its maximum effect at 12 h, which was 10 folds that of the sham group and 1.4 folds that of the I/R group (Figure 2A). As reflected by the increase in MIP2 mRNA levels, a maximal increase of MIP2 protein level was observed in the IPoC group compared with the sham group (4.3 folds) and I/R group (2.1 folds) at 15 h (Figure 2B). These data suggested that IPoC increased MIP2 expression and subsequent MIP2 protein level during the early reperfusion period in the heart.

IPoC induced MIP2 expression in the myocardium. Bar graphs showed the quantitative mRNA expression (A) and protein levels (B) of MIP2 in the myocardium at 1, 3, 6, 9, 12, 15 and 24 h after the reperfusion (n = 3). Values are expressed as ratio versus GAPDH. #P < 0.05 vs sham group; *P < 0.05 vs I/R group. Values are means ± SEM.
MIP2 was up-regulated by HPoC in the in vitro H9c2 myogenic cell model
MIP2 mRNA and protein levels were determined between 1 and 24 h after the reoxygenation in different groups of H9c2 cells. The levels of MIP2 gene expression were markedly increased in the HPoC group compared with the sham group and H/Re group at any time point and reached its peak value at 12 h with a 5.4 folds that of the sham group and 1.6 folds that of the H/Re group (Figure 3A). A significant increase of MIP2 protein was similarly in the HPoC group compared with the sham group and H/Re group at any time point reaching its peak value at 12 h (Figure 3B). Taken together, HPoC increased MIP2 gene and protein expressions in H9c2 myogenic cells.

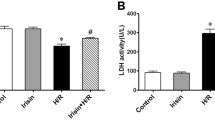
MIP2 involves in the HPoC-induced protection against H/Re mediated LDH release in H9c2 myogenic cells. Bar graphs showed the quantitative mRNA expression (A) and protein levels (B) of MIP2 in the H9c2 myogenic cells at 1, 3, 6, 9, 12, 15 and 24h after the reoxygenation (n = 3). Values are expressed as ratio versus GAPDH, *P < 0.05 vs sham group; #P < 0.05 vs H/Re group. Values are means ± SEM. Transfection with pcDNA3.1-MIP2 and MIP2 siRNA influenced MIP2 protein expression in H9c2 cells (C), MIP2 overexpression and suppression affected HPoC induced protective effect as indicated by LDH release into the medium (D). *P < 0.05 vs the pcDNA3.1 group. #P < 0.05 vs the control siRNA. Values are means ± SEM.
HPoC induced protection of H9c2 cells against apoptosis was enhanced with MIP2 overexpression
After stable transfection with pcDNA3.1-MIP2, the MIP2 protein was overexpressed in the H9c2 cells (Figure 3C). The protective effect of HPoC was further appreciated by decreased level of LDH release in all groups (Figure 3D). Lowest level of LDH activity was observed in the MIP2 overexpressed cells as compared with vector-transfected control cells (Figure 3D). These findings supported the fact that HPoC protected H9c2 cells against death due to H/Re injury.
An elevated peak and the area under the curve of sub-G1 fraction (Figure 4) showed that overexpression of MIP2 significantly inhibited the apoptosis of H9c2 cells as compared with vector-transfected control cells, both in the H/Re group (30.6 ± 1.5% vs. 37.1 ± 2.4%; P < 0.05) and in the HPoC group (22.5 ± 1.9% vs. 30.8 ± 1.5%; P < 0.05). After the study of cell morphology with fluorescent DNA-binding dye Hoechst and propidium iodide (PI), we confirmed that overexpression of MIP2 inhibited the apoptosis (Figure 5) as compared with vector-transfected control cells, either in the H/Re group (36.67 ± 4.41% vs. 52.67 ± 1.45%; P < 0.05) or in the HPoC group (17.67 ± 1.45% vs. 32.67 ± 1.45%; P < 0.05). MIP2 overexpression had no effect on the sham group.

MIP2 involves in the HPoC-induced protection against H/Re mediated H9c2 myogenic cells apoptosis shown by Flow cytometry. Conditions for the treatment groups were as follows: sham group: 15 h of cell culture in normoxic conditions; H/Re group: 6 h of hypoxia and 9 h of reoxygenation; HPoC group: 3 cycles of 10 min reoxygenation and 10 min hypoxia after the 6 h index hypoxia but before the 9 h reoxygenation. *P < 0.05 vs pcDNA3.1 group, #P < 0.05 vs control siRNA group. Values are means ± SEM.

MIP2 involves in the HPoC-induced protection against H/Re mediated H9c2 myogenic cell apoptosis shown by PI and Hoechst staining. Representative pictures showed the morphology of live and dead cells stained by PI (red) and Hoechst (blue) under fluorescent microscope. *P < 0.05 vs pcDNA3.1 group, #P < 0.05 vs control siRNA group. Values are means ± SEM.
HPoC induced protection of H9c2 cells against apoptosis was blunted by MIP2 small interfering RNA (siRNA)
Reduced expression of MIP2 protein was detected in the MIP2 siRNA group compared with the control siRNA group (Figure 3C). LDH release was significantly greater in the MIP2 siRNA group than in the control siRNA group (Figure 3D). As demonstrated by flow cytometry, apoptosis was more prominent in the MIP2 siRNA group compared with control siRNA both in the H/Re group (44.7 ± 1.5% vs. 38.6 ± 0.6%; P < 0.05) and in the HPoC group (36.6 ± 1.1% vs. 31.5 ± 1.0%; P < 0.05) (Figure 4).
Cell apoptosis was characterized by condensed chromatin and fragmented nuclei. The number of apoptotic cells was counted and analyzed. MIP2 RNAi induced a significant increase in cell apoptosis and death as compared with control siRNA, both in the H/Re group (64.0 ± 2.08% vs. 54.0 ± 0.57%; P < 0.05) and in the HPoC group (49.3 ± 2.3% vs. 33.3 ± 0.88%; P < 0.05) (Figure 5). However, MIP2 RNAi did not change the rate of apoptosis in the sham group. MIP2 RNAi blunted the effect of HPoC by reducing the apoptosis of H9c2 cells.
Discussion
From in vivo and in vitro models of ischemia (hypoxia) and reperfusion (reoxygenation), postconditioning has been shown to reduce I/R injury, IPoC reduced the heart infarct size and decreased the serum CK-MB and LDH activity. HPoC significantly reduced the H/Re injury as assessed by decreased LDH release into the medium, and decreased cell apoptosis as shown by flow cytometry and Hoechst/PI staining. This was in agreement with the tissue and cell protection demonstrated from isolated heart and cultured cardiomyocytes in postconditiong (Kin et al., 2005; Sun et al., 2005).
IPoC/HPoC induced MIP2 up-regulation in vivo and in vitro, the release of LDH from the MIP2 overexpression group was lower than that of the MIP2 siRNA group, MIP2 overexpression prevented whereas MIP2 siRNA increased H/Re induced H9c2 cell apoptosis. On the other hand, MIP2 overexpression enhanced HPoC induced protection of H9c2 cells. Suppression of MIP2 expression by RNAi significantly attenuated the HPoC induced anti-apoptotic effect. These findings suggest that endogenous activation of MIP2 represents an attempt in the reparative process by cardiomyocytes. MIP2 may play a role protecting the myocardium cells from being injured by I/R.
The mechanisms of protection by IPoC include: formation and release of several autacoids and cytokines; maintaining acidosis during the early reperfusion; activation of protein kinases; preservation of mitochondrial function, and most strikingly the attenuation of opening the mPTP (Ovize et al., 2010). mPTP is a non-specific pore, permeant to any molecule of < 1.5 kDa, that opens in the inner mitochondrial membrane under oxidative stress and depleted adenine nucleotides. Opening of the mPTP causes massive swelling of mitochondria, rupture of the outer membrane and release of intermembrane components that induce apoptosis (Halestrap et al., 2002). The mPTP opening is a pivotal event in the transition from reversible to irreversible injury during reperfusion. The reduction in infarct size by postconditioning via inhibiting mPTP has been recently reported in in vivo model of ischemia and reperfusion (Tsang et al., 2004).
Recently our data found that MIP2 was localized on the mitochondria, and interacted with voltage-dependent anion channel (VDAC, also known as porin) of mitochondrial outer membrane (data not shown), which is an important component of the mPTP complex. We suppose that the inhibition of mPTP is correlated with a modulation of MIP2 in mitochondria by postconditioning. Further investigation is needed to delineate the relationship between alteration of MIP2 and mPTP opening potentially targeted by postconditioning.
Methods
Surgical procedure and experimental design in rat in vivo model
The rat protocols used in this study were handled in compliance with the guide for the care and use of laboratory animals published by the National Institutes of Health (NIH Publication No. 85-23, Revised 1996). We used intermittent ischemia during the onset of reperfusion after the left coronary artery was occluded as the IPoC model which has been described previously (Kin et al., 2004). In this experiment, healthy male wild-type Sprague-Dawley (SD) rats weighing 280-360 g were anesthetized with 10% chloral hydrate 0.35 ml/100 g intraperitoneally. The animals were ventilated with oxygen-enriched room air using a rodent respirator (50-60 breaths per minute; tidal volume was set to 1.0 ml/100 mg body weight). Rectal temperature was monitored and maintained between 37℃ and 38℃ with surgical table and drapes. A left lateral thoracotomy was performed on the fourth intercostal space and the pericardium opened. A 6-0 proline ligature was placed under the left anterior descending coronary artery (LAD), and the ends of the tie were threaded through a small plastic (PE50) tube to form a snare for reversible LAD occlusion. The rats were randomly assigned to 3 groups based upon the intervention (n = 28 in each group, n = 4 at each time point, Supplemental Data Figure S1A). In the sham group a ligature was placed under the LAD but not tightened. In the I/R and IPoC groups, the LAD was occluded for 45 min. Reperfusion (time t = 0) by loosening the ligature was allowed after 45 min of occlusion in different time courses (1, 3, 6, 9, 12, 15 and 24 h). In the I/R group there was no intervention either before or after the LAD occlusion; In the IPoC group: at the onset of the reperfusion, reflow was initiated with 30 s full coronary flow, followed by 30 s re-occlusion, which was repeated twice for 3 cycles (3 min intervention in total).
Cell culture and transfection
H9c2 myogenic cells were obtained from American Type Culture Collection and were maintained at 37℃ in a humidified atmosphere of 95% air-5% CO2 in Dulbecco's modified Eagle's medium (DMEM, GIBCO) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Hangzhou Sijiqing Biological Technology, China). According to the manufacturer's instructions, a full-length MIP2 cDNA was subcloned into the expression vector pcDNA3.1 (Invitrogen). H9c2 myogenic cells stably expressing MIP2 were established by transfected Lipofectamine 2000 Reagent (Invitrogen) with pcDNA3.1-MIP2. After antibiotic selection (1 mg/ml G418) to eliminate untransfected cells, transfectants were pooled and expanded in 500 µg/ml G418. Parallel cultures of H9c2 myogenic cells were stably transfecting with pcDNA3.1 vector alone. To inhibition the MIP2 expression, RNAs that specifically target the rat MIP2 mRNA were synthesized by Invitrogen (Carlsbad, CA). The small interfering RNA (siRNA) targeting the MIP2 mRNA sequence (UAAAGGCUUUAGUUCAUUCAGG UCA) was found to down-regulate MIP2 expression, as evaluated by Western blot using a specific antibody against total MIP2. Cells were transfected with either MIP2 siRNA or negative control siRNA (TTCTCCGAACGTGTCA CGT) using Lipofectamine 2000 reagent according to the instructions of the manufacturer (Invitrogen).
In vitro protocol
We used cell culture model of HPoC with an in vitro protocol described previously (Sun et al., 2005). After 2-3 days of cell culture in normoxic 10% FBS-DMEM, the culture medium was freshly changed with serum-free DMEM (low glucose). The H9c2 myogenic cells were randomly divided into 3 groups as follows (Supplemental Data Figure S1B): (1) sham: H9c2 myogenic cells were seeded on the dishes and incubated in normoxic conditions (95% air-5% CO2), and the total experimental time course for the other groups was followed; (2) Hypoxia/reoxygenation group (H/Re): the culture dishes were transferred to a hypoxic incubator in a humidified atmosphere that contained 95% N2-5% CO2 for 6 h of hypoxia (PO2 range in the incubator was maintained at < 1%), and subsequently reoxygenated; (3) HPoC group: postconditioning of cells was achieved by incubators (hypoxic and normoxic). At the end of the 6 h hypoxia, the cells were initially transferred to a normoxic incubator for 10 min and then returned to the hypoxic incubator for another 10 min with no change in the culture medium. The HPoC cycle was repeated 3 times and followed by continuous normoxia (i.e. reoxygenation). To reoxygenate, the culture medium in all groups was replaced with fresh oxygenated 10% FBS-DMEM, and the dishes were transferred to a normoxic incubator (95% air-5% CO2) in different time courses (1, 3, 6, 9, 12, 15 and 24 h).
Measurement of infarct size
After the experiment, the heart (n = 5 in each group, reperfusion for 3 h) was immediately removed and frozen at -80℃, and then cut into 7 to 8 transverse slices. The slices were incubated in TTC solution (1%) in PBS at 37℃ for 20 min. The slices were subsequently fixed in 4% formalin solution. The area of the myocardium that was not stained by the TTC was defined as the area of infarction, the risk area being calculated as total ventricular area minus cavities. Infarct size was calculated as percent of the risk area.
Evaluation of tissue injury and cell viability
Lactate dehydrogenase (LDH) and creatine kinase MB (CK-MB) released from cells into blood were used as indicators of myocardial tissue injury in vivo. Serum LDH and CK-MB activities were measured by Olympus AU-640 (Tokyo, Japan). Cell injury of in vitro H9c2 cells was evaluated by an assay of LDH released from the cells into the medium. LDH activity was determined by an LDH assay kit (Jianchen, Nanjing, China), according to the manufacturers' instructions.
Real-time PCR
Total RNA was prepared from the myocardium of rats and H9c2 myogenic cells at each time point using trizol reagent (Invitrogen Life Technologies, Carlsbad, CA), according to the manufacturer's instructions. First-strand cDNA was synthesized with 1 µg of total RNA from each sample and 2 µl of oligo (DT)15 using the Power cDNA Synthesis kit (Promega). 2 µl diluted cDNA (1:10) was amplified with SYBR ®Premix Ex Taq (TaKaRa, Shiga, Japan), in a final volume of 20 µl. The PCR reaction was performed with Applied Biosystems 7300 Real-Time PCR System (USA). The PCR program consisted of initial denaturation at 95℃ for 10 s, 40 cycles at 95℃ for 5 s, and 60℃ for 31 s. The following primers were used: GAPDH forward: 5'-TGCCCCCATGTTTGTGATG-3', GAPDH reverse: 5'-GCTGACAATCTTGAGGGAGTTGT-3'; MIP2 forward: 5'-GTGCTAAGGCATTGTGGTGTGT-3', MIP2 reverse: 5'-GCAACATCT TATCAGTCCATGGTT-3'.
Western blot
The myocardium tissue and H9c2 myogenic cells collected at each time point were lysed in sodium dodecyl sulfate (SDS)-buffer (62 mM Tris-HCl, 1 mM ethylenediamine tetraacetic acid, and 2% SDS, pH 6.8-7.0) containing one tablet of a protease inhibitor cocktail (Complete Mini, Boehringer Mannheim, Germany) per 10 ml of solubilizing buffer, and then incubated for 20 min on ice, and centrifuged at 13000 rpm for 10 min at 4℃. The protein concentration of the supernatant was determined (Bio-Rad Laboratories, Hercules, CA) and 20 µg of the protein was loaded for SDS-polyacrylamide gelelectrophoresis. Proteins were transferred onto PVDF membranes with an electroblotting apparatus. Membranes were blocked for 4 h in Tris-buffered saline (TBS) containing 5% dry milk, incubated with anti-MIP2 polyclonal antibody (Wei et al., 2010) (1:200, Bioss Biological Technology, China) overnight and washed for 3 times (10 min each) with TBS containing 0.1% Tween-20. The membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h each and then washed for 3 times (10 min each) with TBS containing 0.1% Tween-20. Protein bands were visualized with DAB Reagent (Boster Biological Technology, China). Anti-GAPDH antibody (1:1000, Cell Signaling Technology) was used to normalize equal amounts of proteins.
Flow cytometry
Cells were detached from each dish by incubation with trypsin (0.25 mg/ml) and were resuspended in 70% cold ethanol at 4℃ for 18 h after centrifugation. All the cell samples were delivered to Beijing Dingguo Biotechnology for evaluation of the cell cycle progression and death with Coulter Epics XL. Cell death was quantified as a percentage of the sub-G1 peak, which is an indicator of the cell death.
Hoechst/Propidium iodide nuclear staining
Morphology of cells was assessed with Hoechst 33342/PI nuclear staining and viewed by fluorescence microscopy. Cells were harvested, stained with Hoechst and PI, and visualized with a Nikon digital DXM 1200 F camera. A minimum of 200 cells were counted and classified as follows: (I) live cells (normal nuclei: blue chromatin with organized structure); (II) membrane-intact apoptotic cells (bright blue chromatin, which is highly condensed, emarginated, or fragmented); (III) necrotic cells (red, enlarged nuclei with smooth normal structure); (IV) membrane-permeable apoptotic cells (bright red chromatin, highly condensed or fragmented); (V) pyknotic/necrotic cells (dense, bright red, slightly condensed nuclei, sometimes divided into 2 or 3 spheres).
Statistical analysis
Group data are expressed as means ± SEM. Differences between 2 groups were analyzed by unpaired Student's t-test. Differences between three or more groups were analyzed by one-way ANOVA followed by Student-Newman-Keuls test. P < 0.05 was considered statistically significant.
Abbreviations
- CK-MB:
-
creatine kinase MB
- H/Re:
-
Hypoxia/reoxygenation
- HPoC:
-
hypoxia postconditioning
- IPC:
-
ischemic preconditioning
- IPoC:
-
ischemic postconditioning
- I/R:
-
ischemia/reperfusion
- LDH:
-
lactate dehydrogenase
- MIP2:
-
myocardial ischemic preconditioning up-regulated protein 2
References
Berenshtein E, Vaisman B, Goldberg-Langerman C, Kitrossky N, Konijn AM, Chevion M . Roles of ferritin and iron in ischemic preconditioning of the heart . Mol Cell Biochem 2002 ; 234-235 : 283 - 292
Cabrera-Vera TM, Vanhauwe J, Thomas TO, Medkova M, Preininger A, Mazzoni MR, Hamm HE . Insights into G protein structure, function, and regulation . Endocr Rev 2003 ; 24 : 765 - 781
Halestrap AP, McStay GP, Clarke SJ . The permeability transition pore complex: another view . Biochimie 2002 ; 84 : 153 - 166
Hausenloy DJ, Yellon DM . New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway . Cardiovasc Res 2004 ; 61 : 448 - 460
Hausenloy DJ, Tsang A, Yellon DM . The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning . Trends Cardiovasc Med 2005 ; 15 : 69 - 75
Hausenloy DJ, Yellon DM . Survival kinases in ischemic preconditioning and postconditioning . Cardiovasc Res 2006 ; 70 : 240 - 253
Hausenloy DJ, Yellon DM . Preconditioning and postconditioning: new strategies for cardioprotection . Diabetes Obes Metab 2008 ; 10 : 451 - 459
Jiang L, Wang KK, Yuan C, Cheng GW, Liu K, Wang Gl, Xiao XZ . Cloning and characterization of a novel gene . China Journal of Modern Medicine 2008 ; 18 : 2143 - 2145
Joazeiro CA, Hunter T . Biochemistry. Ubiquitination--more than two to tango . Science 2000 ; 289 : 2061 - 2062
Kin H, Zhao ZQ, Sun HY, Wang NP, Corvera JS, Halkos ME, Kerendi F, Guyton RA, Vinten-Johansen J . Postconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting events in the early minutes of reperfusion . Cardiovasc Res 2004 ; 62 : 74 - 85
Kin H, Zatta AJ, Lofye MT, Amerson BS, Halkos ME, Kerendi F, Zhao ZQ, Guyton RA, Headrick JP, Vinten-Johansen J . Postconditioning reduces infarct size via adenosine receptor activation by endogenous adenosine . Cardiovasc Res 2005 ; 67 : 124 - 133
Latchman DS . Heat shock proteins and cardiac protection . Cardiovasc Res 2001 ; 51 : 637 - 646
Murry CE, Jennings RB, Reimer KA . Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium . Circulation 1986 ; 74 : 1124 - 1136
Neer EJ, Schmidt CJ, Nambudripad R, Smith TF . The ancient regulatory-protein family of WD-repeat proteins . Nature 1994 ; 371 : 297 - 300
Ovize M, Baxter GF, Di Lisa F, Ferdinandy P, Garcia-Dorado D, Hausenloy DJ, Heusch G, Vinten-Johansen J, Yellon DM, Schulz R . Postconditioning and protection from reperfusion injury: where do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology . Cardiovasc Res 2010 ; 87 : 406 - 423
Smith TF, Gaitatzes C, Saxena K, Neer EJ . The WD repeat: a common architecture for diverse functions . Trends Biochem Sci 1999 ; 24 : 181 - 185
Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L'Huillier I, Aupetit JF, Bonnefoy E, Finet G, André-Fouet X, Ovize M . Postconditioning the human heart . Circulation 2005 ; 112 : 2143 - 2148
Sun HY, Wang NP, Kerendi F, Halkos M, Kin H, Guyton RA, Vinten-Johansen J, Zhao ZQ . Hypoxic postconditioning reduces cardiomyocyte loss by inhibiting ROS generation and intracellular Ca2+ overload . Am J Physiol Heart Circ Physiol 2005 ; 288 : H1900 - H1908
Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM . Postconditioning: a form of "modified reperfusion" protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway . Circ Res 2004 ; 95 : 230 - 232
van der Voorn L, Ploegh HL . The WD-40 repeat . FEBS Lett 1992 ; 307 : 131 - 134
Vinten-Johansen J . Postconditioning: a mechanical maneuver that triggers biological and molecular cardioprotective responses to reperfusion . Heart Fail Rev 2007 ; 12 : 235 - 244
Wang HC, Zhang HF, Guo WY, Su H, Zhang KR, Li QX, Yan W, Ma XL, Lopez BL, Christopher TA, Gao F . Hypoxic postconditioning enhances the survival and inhibits apoptosis of cardiomyocytes following reoxygenation: role of peroxynitrite formation . Apoptosis 2006 ; 11 : 1453 - 1460
Wei X, Song L, Jiang L, Wang G, Luo X, Zhang B, Xiao X . Overexpression of MIP2, a novel WD-repeat protein, promotes proliferation of H9c2 cells . Biochem Biophys Res Commun 2010 ; 393 : 860 - 863
Yellon DM, Hausenloy DJ . Myocardial reperfusion injury . N Engl J Med 2007 ; 357 : 1121 - 1135
Yu L, Gaitatzes C, Neer E, Smith TF . Thirty-plus functional families from a single motif . Protein Sci 2000 ; 9 : 2470 - 2476
Yuan C, Zhang HL, Liu Y, Xiao XZ . Cloning and Characterization of new genes MIP21 and HMIP22 up-regulated in myocardial ischemic preconditioning . Chin J Arterioscler 2003 ; 11 : 86 -
Zhao J, Liu Y, Wei X, Yuan C, Yuan X, Xiao X . A novel WD-40 repeat protein WDR26 suppresses H2O2-induced cell death in neural cells . Neurosci Lett 2009 ; 460 : 66 - 71
Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J . Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning . Am J Physiol Heart Circ Physiol 2003 ; 285 : H579 - H588
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (46987001) and Major National Basic Research Program of China (No. 2007CB-512007).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhu, HL., Wei, X., Qu, SL. et al. Ischemic postconditioning protects cardiomyocytes against ischemia/reperfusion injury by inducing MIP2. Exp Mol Med 43, 437–445 (2011). https://doi.org/10.3858/emm.2011.43.8.049
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2011.43.8.049
Keywords
This article is cited by
-
MiR-144-5p/CCL12 Signaling Axis Modulates Ischemic Preconditioning-Mediated Cardio-protection by Reducing Cell Viability, Enhancing Cell Apoptosis, Fibrosis, and Pyroptosis
Applied Biochemistry and Biotechnology (2023)
-
Differentiation-Dependent Doxorubicin Toxicity on H9c2 Cardiomyoblasts
Cardiovascular Toxicology (2012)
