
Abstract
To examine copy number variations among the Korean population, we compared individual genomes with the Korean reference genome assembly using the publicly available Korean HapMap SNP 50 k chip data from 90 individuals. Korean individuals exhibited 123 copy number variation regions (CNVRs) covering 27.2 mb, equivalent to 1.0% of the genome in the copy number variation (CNV) analysis using the combined criteria of P value (P < 0.01) and standard deviation of copy numbers (SD ≥ 0.25) among study subjects. In contrast, when compared to the Affymetrix reference genome assembly from multiple ethnic groups, considerably more CNVRs (n = 643) were detected in larger proportions (5.0%) of the genome covering 135.1 mb even by more stringent criteria (P < 0.001 and SD ≥ 0.25), reflecting ethnic diversity of structural variations between Korean and other populations. Some CNVRs were validated by the quantitative multiplex PCR of short fluorescent fragment (QMPSF) method, and then copy number invariant regions were detected among the study subjects. These copy number invariant regions would be used as good internal controls for further CNV studies. Lastly, we demonstrated that the CNV information could stratify even a single ethnic population with a proper reference genome assembly from multiple heterogeneous populations.
Similar content being viewed by others

Introduction
Human genetic variations comprise various types of structural genomic changes and single nucleotide polymorphisms (SNPs). Large microscopic changes affect more than tens of millions of bases (mb) in the genome, and are rare in healthy individuals, but smaller structural variations ranging from 1 kb to hundreds of kb are frequent and widespread even in normal individuals, contributing to human genetic diversity or disease susceptibility (Feuk et al., 2006; Freeman et al., 2006). Such submicroscopic genomic variations have been defined in terms of copy number variations (CNVs) and include large-scale copy number variants (LCVs) (Iafrate et al., 2004), copy number polymorphisms (CNPs) (Sebat et al., 2004), and intermediate-sized variants (ISVs) (Tuzun et al., 2005), as well as other types of genomic variations such as low copy repeats (LCRs) (Lupski and Stankiewicz, 2005), multisite variants (MSVs) (Fredman et al., 2004), and paralogous sequence variants (PSV) (Eichler 2001). However, by convention, genomic variations do not include variants that arise from the insertion/deletion of transposable elements (Freeman et al., 2006).
Various experimental platforms and analytical tools such as array-based methods (SNP genotyping array, BAC- and oligonucleotide-array CGH) and clone-based large scale sequencing approaches have been utilized to study structural genomic variations in humans as well as other species (Li et al., 2004; Newman et al., 2005; Perry et al., 2006; Dumas et al., 2007; Graubet et al., 2007; Human Genome Structural Variation Working Group 2007). In humans, multiple studies, including the international HapMap project, have so far annotated CNVs to more than 4000 distinct regions spanning 600 mb, though their abundance and size are likely to be overestimated due to variability of methods and fewer cross-platform validations (Cooper et al., 2007). Since most CNV detection technologies rely on a comparison to a reference genome, CNVs are determined when cross-referenced to disease-affected individuals or different ethnic populations (Rodriguez-Revenga et al., 2007). Thus, absolute copy number information, which is especially important for clinical diagnosis or assessment of disease susceptibility, cannot be easily determined by current quantitative assays except Fiber-FISH. Nonetheless, recent studies have shown that DNA copy number variations are implicated in human diseases including glomerulonephritis (FCGR3B) (Aitman et al., 2006), HIV-1/AIDS (CCL3L1) (Gonzalez et al., 2005), bipolar disorder and schizophrenia (GLUR7, CACNG2 and AKAP5) (Wilson et al., 2006), muscular atrophy (SMN) (Kesari et al., 2005) and neoplasia (14q12) (Braude et al., 2006).
On the other hand, recent reports also suggest that different ethnic groups may represent different profiles of CNVs that are stratified in the human population (Redon et al., 2006; Kidd et al., 2007). In our previous BAC array CGH study, Korean copy number variants were discovered when compared to reference DNA from different ethnic groups (Jeon et al., 2007). In an attempt to obtain a standard CNV profile for the Korean population, which would facilitate association studies of CNVs with disease susceptibility as well as population genetic diversity, we analyzed a comprehensive CNV profile of 90 Korean individuals using the publicly available Korean HapMap SNP 50 k chip data sets and tested its application to population stratification.
Results
To generate CNV profiles of Koreans, we extracted DNA copy number information from the publicly available Korean HapMap SNP 50 k chip data (http://www.khapma.org), and then conducted either P value-based or copy number-based CNV analyses as well as the combined P value and copy number-based CNV analysis using two different copy number reference genome assembly sets. Basically, two different reference sets were used to detect CNVs from study subjects (n = 90); 1) the Korean reference set generated from all the genomes of 90 individuals, 2) the Affymetrix reference set provided as a copy number reference from multiple ethnic groups by Affymetrix Inc. We tested the validity of different CNV calling criteria by the quantitative multiplex PCR of short fluorescent fragment (QMPSF) experiments. The best validation rate was observed in the combined CNV calls with P and SD values.
P value-based CNV analysis (cutoff P < 0.01 or P < 0.001)
Our P value-based CNV analysis using the Korean reference set showed that 90 Korean individuals represented 435 copy number variation regions (CNVRs) covering 123 mb equivalent to 4.1% of the genome using a cutoff of P < 0.01, while the choice of a more stringent cutoff of P < 0.001 allowed detection of less CNVRs (n = 126) covering 35 mb (1.2%) (Supplemental Data Table S1). In contrast, when the Affymetrix reference set from multiple ethnic groups was used to detect CNVRs from Korean individuals, the more stringent cutoff of P < 0.001 was chosed because this cutoff of P < 0.001 provided enough stringency in CNV calling to get a CNV profile of a reasonable number of CNVRs. Indeed, even stringent criteria of CNV calling detected more CNVRs (n = 2034) covering 594 mb equivalent to 19.8% of the genome (Supplemental Data Table S1). The proportion of CNVRs on a given chromosome varies from 11.3% on chromosome 14 to 44% on chromosome 12, with the mean proportion of 19.8% on average for all chromosomes. Our P value-based CNV analysis using the Affymetrix reference set (P < 0.001) showed that CNVRs were uniformly distributed across the human chromosomes, and the population-wide occurrence of particular CNVRs ranged from zero to 72 out of 90 individuals (data not shown). According to the results of QMPSF experiments for the CNV calls detected by P value-based CNV analysis using the Korean reference set, the validation rate was approximately 50% (3 out of 6 CNVRs) of tested CNVRs (Table 1, see also Supplemental Data Materials for CNV validation).
Copy number-based CNV analysis (cutoff SD ≥ 0.25)
We also employed the standard deviation (SD ≥ 0.25) of copy numbers of each probe for the 90 individuals as the criteria of CNV calling in the copy number-based CNV analysis, which detected the population-wide CNVRs among the Korean population. This copy number-based CNV analysis detected 595 CNVRs (8.9% of the genome) and 790 CNVRs (11.8%) from 90 individuals, using the Korean reference and the Affymetrix reference sets, respectively (Supplemental Data Table S1). The average length of CNVRs was approximately 448kb when using both reference genomes. The validation rate was approximately 46% (18 out of 39 CNVRs) of tested CNVRs (Table 1, and see also Supplemental Data Materials for CNV validation).
Combined CNV analysis with P value (P < 0.01 or 0.001) and copy numbers (SD ≥ 0.25)
When compared with the Korean reference set using the combined criteria of P value (P < 0.01) and standard deviation of copy numbers (SD ≥ 0.25) of given probes among study subjects, Korean individuals (n = 90) exhibited 123 CNV regions (CNVRs) encompassing 27.2 mb, equivalent to 1.0% of the genome (Table 2, and see also Supplemental Data Table S4 for CNVR list). In contrast, when compared with the Affymetrix reference set, the combined CNV analysis (P < 0.001 and SD ≥ 0.25) detected more CNVRs (n = 643) encompassing 135.1 mb in larger proportions (5.0%) of the genome (Table 2, and see also Supplemental Data Table S5 for CNVR list). The proportion of copy number gains was lower than that of copy number losses when compared with the Korean reference set, whereas the ratio of gains to losses was higher when compared with the Affymetrix reference set (Figure 1).

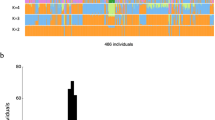
Composition of population-wide CNVRs. (A) Population-wide CNVRs were defined for two or more consecutive probes among study subjects (n = 90) using the combined CNV analysis of P and SD values. High or low copy numbers of given genomic regions in relative to a reference genome assembly set were classified into gain or loss of CNVRs. The mixed type of CNVRs was defined when a genomic region encompassing two or more consecutive probes represented both types of gain and loss of CNVRs among the study population. (B) Composition of CNVR types. CNVRs were detected by the combined CNV analysis using the Korean and Affymetrix reference sets, and then were classified into three CNVR types (gains, losses, or mixed types).
Standard deviation of copy numbers allowed us to detect only population-wide CNVRs among the study subjects (n = 90) which could not provide the number of CNVRs per person, while the P value-based CNV analysis could detect individual-based CNVRs. Therefore, the combined criteria of P and SD values could detect both population-wide and individual-based CNVRs, enhancing the reliability of CNV calls. Indeed, the validation rate was approximately 75% (9 out of 12 CNVRs) of tested CNVRs, which was higher than those of other CNV calls (Table 1 and Supplemental Data Materials for CNV validation).
Properties of Korean CNVs
According to the results of the combined CNV analysis, an average number of CNVRs per person was 2.1 ± 5.0 (ranging from 0 to 32 CNVRs) when compared to the Korean reference set (Figure 2). Thirteen individuals exhibited 5 or more CNVRs, and the top three highest numbers of CNVRs were detected in individuals of KR-41 (32 CNVRs), KR-72 (27 CNVRs) and KR-70 (15 CNVRs). Fifty individuals did not represent CNVRs when compared to the Korean reference set using the combined criteria of P and SD values (Figure 2).

Numbers of CNVRs per person. The CNVRs were detected by the combined CNV analysis with P and SD values using the Korean and Affymetrix reference set. The average number of CNVRs per person was 2.1 ± 5.0 (mean ± STD) (up to 32 CNVRs at KR-41 sample) when compared with the Korean reference set, and 33.1 ± 26.6 (mean ± STD) (up to 124 CNVRs at KR-41 sample) when compared with the Affymetrix reference set.
In contrast to the Korean reference set, the Affymetrix reference set allowed detection of more CNVRs in the combined CNV analysis (P < 0.001 and SD ≥ 0.25). An average number of CNVRs per person was 33.1 ± 26.6 (ranging from 2 to 124 CNVRs) (Figure 2). Thus, the average number of CNVRs per person was much higher in the combined CNV analysis using the Affymetrix reference than the Korean reference. This observation may be ascribed to ethnic diversity between the Korean reference set from a single population and the Affymetrix reference set from the multiple ethnic groups. On the other hand, KR-2 and KR-4 samples represented lowest numbers of CNVRs when referenced to both the Korean and Affymetrix reference sets, while KR-41 and KR-72 samples represented highest numbers of CNVRs. The average length of individual-based CNVRs exhibited no big difference in the combined CNV analysis between the Korean reference set (128 kb per CNVR) and the Affymetrix reference set (142 kb per CNVR).
The most frequent CNVR (KC16-T01) was detected in 7 individuals (7.8%) out of the study subjects (n = 90) in the combined CNV analysis using the Korean reference set (Supplemental Data Table S4). Seventy CNVRs (57%) of 123 CNVRs in total were occurred in single individuals while 53 CNVRs (43%) were occurred in two or more individuals (7 individuals at most). The most frequent CNVR (KC16-T01) was localized in 16p12.1 with a higher occurrence (n = 7) than that of a CNVR (n = 4) at the Ig locus, suggesting that this frequent CNVR may be one of highly susceptible CNV targets as much as Ig loci.
Chromosome 21 and 18 represented the least proportion (0.03% and 1.86% of the corresponding chromosome) of CNVRs using the Korean reference set and the Affymetrix reference set, respectively (Table 2). This observation suggests that chromosome 21 may be relatively conserved for genomic structural variations within the Korean population. When the two criteria (P < 0.01 and SD ≥ 0.25) were combined in CNV calling using the Korean reference set, 64 CNVRs (52.1%) of 123 CNVRs were known while 59 CNVRs (47.9%) were unknown according to the Database of Genomic Variants (http://projects.tcag.ca/variation) (Supplemental Data Table S4).
Identification of copy number invariant regions and qPCR validation of CNVRs
Based on the result of copy number-based CNV analysis, we first selected eleven copy number invariant regions showing the lowest standard deviation (SD) of GSA_CN values (Table 3). These regions exhibited almost no variation in DNA copy numbers among all individuals when compared to the self-including Korean reference set. One of them (2q36.1) was validated for no copy number variation in reference to copy numbers of Factor VIII gene among all study subjects (n = 90) (Figure 3).

Validation of copy number invariant regions. DNA copy numbers of a copy number invariant region (CN2-2) at 2q36.1 was quantitated for all study subjects (n = 90) by the QMPSF method. Relative copy numbers of CN2-2 to Factor VIII was 1.32 ± 0.08 with the coefficient of variation (6.3%) among the test subjects. Error bars indicate standard deviations from three independent QMPSF measurements performed in triplicate.
Next, we evaluated the reliability of CNV calls from P value-based and copy number-based analyses, as well as the combined P value and copy number-based analysis in order to determine the better CNV calling rule. Since our Korean reference set was generated from averaged copy numbers of a given probe for all of 90 individuals, the coefficient of variation (CV) was used as a statistical criteria of copy number difference among the tested subjects (n = 90). Therefore, when a particular CNVR displayed over 8% of the coefficient of variation of copy number measurements from the QMPSF results, we considered the particular CNVR to be validated. As a result, the validation rate of CNVRs was higher in the combined CNV analysis than P value-based or copy number-based CNV analyses (Table 1, and Supplemental Data Materials for CNV validation).
Refining the population stratification of 90 individuals
CNV profiles identified in this study were applied to the principle component analysis to stratify the 90 individuals. Multi-dimensional scaling showed that the first and second eigenvectors explained 21% and 9% of the distance variables, respectively (Figure 4). When referenced to the Affymetrix reference, the 90 individuals were dispersed in a plot of eigenvetors, suggesting that the Korean population can be stratified into subgroups according to DNA copy number variations. In contrast, most of the 90 individuals were centered around an intersecting point of two eigenvectors when referenced to the self-including Korean reference genome. This result suggests that a reference genome assembly from the more heterogeneous population gives more informative data for population stratification studies using CNV information. Thus, if an appropriate copy number reference genome is selected, genomic structural variation would provide a valuable source for refining stratification of ethnic groups within a single population.

Plots of two eigenvectors for 90 unrelated individuals from the P value-based analysis of DNA copy numbers. The first and second eigenvectors explain 21% and 9% of the distance variables, respectively. The Affymetrix reference that was generated from more heterogeneous populations resulted in a more widely dispersed plot than the self-including Korean reference that was generated from a single ethnic population.
Discussion
We analyzed the profiles of DNA CNVs of 90 Korean individuals using publicly available Korean HapMap SNP 50k chip data. Different CNV reference genome assembly sets (either the Korean reference or Affymetrix reference sets) were used for the combined CNV analysis with P and SD values, as well as P value-based and copy number-based analyses. As results of QMPSF experiments, the validation rate of CNVRs was higher in the combined CNV analysis than P value-based or copy number-based CNV analyses. Thus, here we finally generated the CNV profile of a Korean population by the combined CNV analysis. The Korean HapMap samples (n = 90) represented five times less CNVRs in total when referenced to the Korean reference set (123 CNVRs) than the Affymetrix reference set (643 CNVRs), reflecting an ethnic difference in CNV profiles between the Korean population and other ethnic groups. In fact, it was reported that copy numbers of particular genes (e.g., CCL3L1, MAPT) varied among different ethnic groups (Gonzalez et al., 2005). Thus, this result suggests that each ethnic population has a distinct CNV profile applicable to various population genetic studies.
On the other hand, in the combined CNV analysis with both P and SD values, no CNVR was detected in 50 individuals when referenced to the self-including Korean reference set. These 50 individuals would represent the standard genome of the Korean population in terms of structural genomic variations because they did not exhibit a copy number difference in relative to the Korean reference genome. Thus, genetic information of these 50 individuals could be a good standard reference for further population genetic studies.
Some array CGH studies detected 11 CNVs (Sebat et al., 2004) or 12.4 CNVs (Iafrate et al., 2004) on average in each person. Generally, array CGH assays identifies a smaller number of CNVRs comprising large-insert clones, which result in the overestimation of CNV length (Redon et al., 2006). Moreover, the different choice of reference genome sets can affect total and average numbers of CNVRs in normal healthy individuals. In this study, the Korean population exhibited 123 CNVRs in total and 2.1 ± 5.0 per person (up to 32 CNVRs) when compared with the Korean reference genome set in the combined CNV analysis with cutoffs of P < 0.01 and SD ≥ 0.25. These CNV calls were made in reference to the self-including Korean genome assembly which was generated from the average copy numbers for the study subjects. Therefore, the choice of self-including reference genome assembly set might contribute to small numbers of CNV calls.
Generally, the term polymorphic is used to denote variants that occur in > 1% of the population (Strachen and Read, 1999). CNVRs identified in this study are considered to be potentially polymorphic rather than rare mutations in the Korean population, because those CNVRs were detected at least once more in 90 individuals. Furthermore, these structural genomic variations provide an additional dimension for human genetic diversity and disease susceptibility along with SNPs. Thus, it would be interesting to study the simultaneous association of these potentially polymorphic CNVRs and SNPs of particular genes with human complex diseases including diabetes and hypertension.
Many CNV analysis tools have been developed to extract accurate copy number information from probe intensities of SNP genotyping arrays (Lin et al., 2004; Nannya et al., 2005; Price et al., 2005; Slater et al., 2005; Fiegler et al., 2006; Hu et al., 2006). However, the detection of real CNVs still needs additional experiments for validation, such as PCR-based methods (e.g. quantitative real-time PCR, QMPSF, MAPH, MLPA) and hybridization-based methods (e.g., Fiber-FISH, Southern blotting) (Feuk et al., 2006). According to the QMPSF results, the validation rates of CNV calls from our different CNV analyses were 50%, 46% and 75% for P value-based, copy number-based analysis, and the combined CNV analysis with P and SD values, respectively. The P value-based CNV analysis could detect individual-based CNVRs whereas the copy number-based CNV analysis with SD values could detect population-wide CNVRs. Thus, our combined CNV analysis enabled to detect population-wide as well as invidividual-based CNVRs with the higher validation rate (75%) of CNV calls. Redon group estimated false positive rate using singleton CNVs (called in only a single individual) (Redon et al., 2006). They found an initial validation failure rate of 24% and then finally estimated a false positive rate of 8% by extrapolating these validation rates across the entire data set (24% multiplied by the frequency of singleton CNVs called on only one plate form).
There are possible explanations for low validation rates of our CNV calls. First, our CNV calls were made in reference to the genome assembly set from multiple individuals (n = 90) instead of the single genome, which could not appropriately provide a single reference DNA sample for QMPSF experiments. Second, QMPSF primers might not exactly target authentic CNV regions because our CNV profile was generated from the 50 k Affymetrix chip data which contained a relatively low density of SNP probes, contributing to detect a low resolution of CNVRs in the present study. Third, an alternative explanation is that some CNVRs might be multi-allelic or complex CNVRs that were difficult to be validated by conventional PCR based methods. Rather, hybridization based methods (e.g., Fiber-FISH, Southern blotting) or a whole genome sequencing approach would detect such multi-allelic or complex CNVRs (Rodriguez-Revenga et al., 2007). Taken together, the validation rate would be increased if CNV-affected samples were compared with a single genome reference in validation experiments of CNVRs. Moreover, more advanced chip platforms with a higher density of probes would give a higher-resolution map of CNVRs, which supports better experimental validation.
The longest CNVR detected by P value-based CNV anlaysis was rather a microscopic genome change encompassing approximately 31 mb at 4q13.14q22.13, which was validated in the corresponding EBV-transformed lymphoblastoid cell line (LCL) by the QMPSF method. The copy number change of this region was not found in blood DNA from the same donor as the LCL DNA (data not shown), suggesting that this copy number change was acquired during EBV-mediated B cell transformation. The LCL strain containing this CNVR would provide a valuable resource for a haploinsufficiency study of corresponding genes at this locus.
An immunoglobulin locus (22q11.22) was also detected as CNV-affected regions with the occurrence of four (4/90) in the combined CNV analysis using the Korean reference set. Previous reports suggested that polymorphic gene duplication may frequently occur in Ig loci including 14q32.33 (Ig heavy variable cluster) and 22q11.2 (Ig heavy chain constant region and Ig lamda) (Sasso et al., 1995; van der Burg et al., 2002; Buckland et al., 2003). It is likely that the copy number variation of the Ig loci may not be from the germ line but a de novo CNV enriched during LCL generation. In fact, Ig locus CNVs were excluded in CNV analyses (Redon et al., 2006).
Recent reports suggest that as much as 40% of the known CNVs occur in gene deserts while the other CNVs are enriched for genes involved in immunity and environmental responses (Derti et al., 2006; Rodriguez-Revenga et al., 2007). Copy number invariant regions may be evolutionary conserved for DNA copy numbers because of the potential effects of gene dosage. In addition, with respect to genome stability, it would be interesting to know whether individuals who contain more CNVRs have a higher risk of cancer than individuals who contain less CNVRs. Therefore, CNVRs and non-CNVRs identified in this study would be a good starting point of further CNV studies to determine their clinical relevance in complex traits or disease susceptibility in the Korean population.
Methods
DNA samples
We used the Affymetrix GeneChip Mapping 50 k array data set obtained from the Korean HapMap project for which DNA samples were selected to include 90 unrelated healthy Korean individuals with an equal sex ratio and age 40-69 years for the Korean Health and Genome Epidemiology Study (http://cgs.cdc.go.kr), as described in previous reports (Kim et al., 2006; Yoo et al., 2006). Genomic DNA was extracted from EBV-transformed B lymphoblastoid cell lines (LCLs) provided by the National Biobank of Korea, Korea National Institute of Health.
DNA copy number analysis
The Affymetrix GeneChip Mapping 50k_XBA240 array data for the Korean HapMap project (Korean HapMap 50 k) were generated according to manufacturer's instructions, as reported elsewhere (Herbert et al., 2006). Average call rate of the Korean HapMap 50 k data set was 98.4% with a standard deviation of 0.92%, ranging from 95.1% to 99.4%. For CNV detection, the Korean HapMap 50 k data were analyzed using Affymetrix GeneChip Chromosome Copy Number Analysis Tool (CNAT) 3.0. DNA copy numbers of individual Koreans (n = 90) were compared to two different copy number reference genome assembly sets: 1) the Affymetrix Mapping 50k_XBA240 reference data (hereinafter called the Affymetrix reference set) from three different ethnic groups (42 African Americans, 20 Asians, and 42 Caucasians) and the fourth group (24 PD panel) in which the 20 Asians did not include Koreans, 2) the self-including copy number reference genome assembly from 90 Korean individuals of the Korean HapMap 50 k (hereinafter called the Korean reference set).
With regard to CNV calling rules, we annotated CNVRs to particular chromosomal regions if the region was more than 1kb in size, encompassed two or more consecutive probes, and met either of the following cutoff criteria: 1) P < 0.001 from the GSA_pVal (genome-smoothed analysis of the P-value) when compared with the Affymetrix reference, 2) P < 0.01 from the GSA_pVal when compared with the self-including Korean reference, 3) a standard deviation (SD ≥ 0.25) of GSA_CN values (genome-smoothed analysis of the copy number) in copy number-based analysis, 4) the combined criteria of P value (P < 0.01 or P < 0.001) and standard deviation (SD ≥ 0.25) of copy numbers. DNA copy number variations of the sex chromosomes were not analyzed in this study because of different sex ratios of test samples to reference genome assemblies.
CNV validation
DNA copy numbers were validated by the QMPSF method using fluorescein-labeled forward primers, as described before (Charbonnier et al., 2002; Vaurs-Barriere et al., 2006). Briefly, 100 ng of DNA template was added to make 25 µl of PCR reaction mixture including 1× PCR Gold buffer, 2 mM MgCl2, 0.2 mM dNTP, appropriate PCR primers at concentrations of 0.04-0.09 µM, and 3 units of Ampli-Taq Gold (Applied Biosystems). One of copy number invariant regions identified among the 90 individuals in this study (CN2-2) or the coagulation factor VIII gene was used as an internal reference DNA for the normalization of input DNA (Levine et al., 2005; Jeon et al., 2007). Primer sequences of reference genes were as follows. CN2-2: forward, 5'-CTTAGGTTCCCACGGTTTGA-3'; reverse 5'-GCACTTGAAAGGTGCCTAGC-3', Factor VIII: forward, 5'-TACCATCCAGGCTGAGGTTTAT-3', reverse, 5'-AAAGAGTTGTAACGCCACCATT-3'. Hot start PCR was carried out at 95℃ for 10 min for a denaturation step, followed by 21 cycles of 94℃ for 30 s, 60℃ for 30 s, and then further extended at 72℃ for 50 min. Ethanol precipitated PCR products were dissolved in 5 µl of water. One microliter of the purified PCR products was mixed with 1 µl of Gene Scan-500 LIZ size standards and 14 µl of HiDi formamide, and then run on the ABI3730 capillary sequencer (Applied Biosystems). Data analysis was performed using the GeneMapper software (Applied Biosystems). In order to obtain experimental copy numbers of CNVRs, the relative peak height of a given CNVR was devided by the peak height of a reference of Factor VIII or CN2-2 from the QMPSF chromatogram, which resulted in relative copy numbers of CNVRs (see Supplemental Data Materials for QMPSF experiments in details).
Multi-dimensional scaling (MDS)
After CNV profiles of the 90 individuals were obtained by the P value-based analysis in reference to the Affymetrix (P < 0.001) or Korean (P < 0.01) reference genome assembly sets, loss and gain of DNA copy numbers were coded to "1" and "2" for each probe, respectively. Probes with no copy number change were coded to zero "0". These codes of copy number patterns were used in principle component analysis. Briefly, the pairwise similarity coefficients (sij) of CNV for individuals were computed as;

where cijk is 1 (or 0) depending on the copy number change (or no change) between two individuals (i and j) at a marker position(k), N is the number of markers and L (=M) is the number of subjects. In particular, the diagonal matrix elements (sii) are not normalized to be the same among individuals since the number of accumulated copy number changes in a whole genome could be different individually. Using the similarity coefficient (sij) Jacobi transformation was performed to calculate eigenvectors (Press et al., 1988).
Abbreviations
- CNVs:
-
copy number variations
- CNVR:
-
copy number variation region
- LCL:
-
lymphoblastoid cell line
- QMPSF:
-
quantitative multiplex PCR of short fluorescent fragment
References
Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, Mangion J, Roberton-Lowe C, Marshall AJ, Petretto E, Hodges MD, Bhangal G, Patel SG, Sheehan-Rooney K, Duda M, Cook PR, Evans DJ, Domin J, Flint J, Boyle JJ, Pusey CD, Cook HT . Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans . Nature 2006 ; 439 : 851 - 855
Braude I, Vukovic B, Prasad M, Marrano P, Turley S, Barber D, Zielenska M, Squire JA . Large scale copy number variation (CNV) at 14q12 is associated with the presence of genomic abnormalities in neoplasia. 1 . BMC Genomics 2006 ; 7 : 138 -
Buckland PR . Polymorphically duplicated genes: their relevance to phenotypic variation in humans . Ann Med 2003 ; 35 : 308 - 315
Charbonnier F, Olschwang S, Wang Q, Boisson C, Martin C, Buisine MP, Puisieux A, Frebourg T . MSH2 in contrast to MLH1 and MSH6 is frequently inactivated by exonic and promoter rearrangements in hereditary nonpolyposis colorectal cancer . Cancer Res 2002 ; 62 : 848 - 853
Cooper GM, Nickerson DA, Eichler EE . Mutational and selective effects on copy-number variants in the human genome . Nat Genet 2007 ; 39 : S22 - S29
Derti A, Roth FP, Church GM, Wu CT . Mammalian ultraconserved elements are strongly depleted among segmental duplications and copy number variants . Nat Genet 2006 ; 38 : 1216 - 1220
Dumas L, Kim YH, Karimpour-Fard A, Cox M, Hopkins J, Pollack JR, Sikela JM . Gene copy number variation spanning 60 million years of human and primate evolution . Genome Res 2007 ; 17 : 1266 - 1277
Eichler EE . Recent duplication, domain accretion and the dynamic mutation of the human genome . Trends Genet 2001 ; 17 : 661 - 669
Feuk L, Carson AR, Scherer SW . Structural variation in the human genome . Nat Rev Genet 2006 ; 7 : 85 - 97
Fiegler H, Redon R, Andrews D, Scott C, Andrews R, Carder C, Clark R, Dovey O, Ellis P, Feuk L, French L, Hunt P, Kalaitzopoulos D, Larkin J, Montgomery L, Perry GH, Plumb BW, Porter K, Rigby RE, Rigler D, Valsesia A, Langford C, Humphray SJ, Scherer SW, Lee C, Hurles ME, Carter NP . Accurate and reliable high throughput detection of copy number variation in the human genome . Genome Res 2006 ; 16 : 1566 - 1574
Fredman D, White SJ, Potter S, Eichler EE, Den Dunnen JT, Brookes AJ . Complex SNP-related sequence variation in segmental genome duplications . Nat Genet 2004 ; 36 : 861 - 866
Freeman JL, Perry GH, Feuk L, Redon R, McCarroll SA, Altshuler DM, Aburatani H, Jones KW, Tyler-Smith C, Hurles ME, Carter NP, Scherer SW, Lee C . Copy number variation: new insights in genome diversity . Genome Res 2006 ; 16 : 949 - 961
Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, Murthy KK, Rovin BH, Bradley W, Clark RA, Anderson SA, O'connell RJ, Agan BK, Ahuja SS, Bologna R, Sen L, Dolan MJ, Ahuja SK . The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility . Science 2005 ; 307 : 1434 - 1440
Graubert TA, Cahan P, Edwin D, Selzer RR, Richmond TA, Eis PS, Shannon WD, Li X, McLeod HL, Cheverud JM, Ley TJ . A high-resolution map of segmental DNA copy number variation in the mouse genome . PLoS Genet 2007 ; 3 : e3 -
Herbert A, Gerry NP, McQueen MB, Heid IM, Pfeufer A, Illig T, Wichmann HE, Meitinger T, Hunter D, Hu FB, Colditz G, Hinney A, Hebebrand J, Koberwitz K, Zhu X, Cooper R, Ardlie K, Lyon H, Hirschhorn JN, Laird NM, Lenburg ME, Lange C, Christman MF . A common genetic variant is associated with adult and childhood obesity . Science 2006 ; 312 : 279 - 283
Hu N, Wang C, Hu Y, Yang HH, Kong LH, Lu N, Su H, Wang QH, Golstein AM, Buetow KH, Emmert-Buck MR, Taylor PR, Lee MP . Genome-wide loss of heterozygosity and copy number alteration in esophageal squamous cell carcinoma using the Affymetirx GeneChip Mapping 10K array . BMC Genomics 2006 ; 7 : 1 - 16
Eichler EE, Nickerson DA, Altshuler D, Bowcock AM, Brooks LD, Carter NP, Church DM, Felsenfeld A, Guyer M, Lee C, Lupski JR, Mullikin JC, Pritchard JK, Sebat J, Sherry ST, Smith D, Valle D, Waterston RH . Completing the map of human genetic variation . Nature 2007 ; 447 : 161 - 165
Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C . Detection of large-scale variation in the human genome . Nat Genet 2004 ; 36 : 949 - 951
Jeon JP, Shim SM, Nam HY, Baik SY, Kim JW, Han BG . Copy number increase of 1p36.33 and mitochondrial genome amplification in Epstein-Barr virus-transformed lymphoblastoid cell lines . Cancer Genet Cytogenet 2007 ; 173 : 122 - 130
Kesari A, Idris MM, Chandak GR, Mittal B . Genotype-phenotype correlation of SMN locus genes in spinal muscular atrophy patients from India . Exp Mol Med 2005 ; 37 : 147 - 154
Kidd JM, Newman TL, Tuzun E, Kaul R, Eichler EE . Population stratification of a common APOBEC gene deletion polymorphism . PLoS Genet 2007 ; 3 : e63 -
Kim KJ, Lee HJ, Park MH, Cha SH, Kim KS, Kim HT, Kimm K, Oh B, Lee JY . SNP identification, linkage disequilibrium, and haplotype analysis for a 200-kb genomic region in a Korean population . Genomics 2006 ; 88 : 535 - 540
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D'Andrea A, Frohling S, Dohner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG . Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis . Cancer Cell 2005 ; 7 : 387 - 397
Li J, Jiang T, Mao JH, Balmain A, Peterson L, Harris C, Rao PH, Havlak P, Gibbs R, Cai WW . Genomic segmental polymorphisms in inbred mouse strains . Nat Genet 2004 ; 36 : 952 - 954
Lin M, Wei LJ, Sellers WR, Lieberfarb M, Wong WH, Li C . dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data . Bioinformatics 2004 ; 20 : 1233 - 1240
Lupski JR, Stankiewicz P . Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes . PLoS Genet 2005 ; 1 : e49 -
Nannya Y, Sanada M, Nakazaki K, Hosoya N, Wang L, Hangaishi A, Kurokawa M, Chiba S, Bailey DK, Kennedy GC, Ogawa S . A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays . Cancer Res 2005 ; 65 : 6071 - 6079
Newman TL, Tuzun E, Morrison VA, Hayden KE, Ventura M, McGrath SD, Rocchi M, Eichler EE . A genome-wide survey of structural variation between human and chimpanzee . Genome Res 2005 ; 15 : 1344 - 1356
Perry GH, Tchinda J, McGrath SD, Zhang J, Picker SR, Cacers AM, Iafrate AJ, Tyler-Smith C, Scherer SW, Eichler EE, Stone AC, Lee C . Hotspots for copy number variation in chimpanzees and humans . Proc Natl Acad Sci USA 2006 ; 103 : 8006 - 8011
Press WH, Flannery BP, Teukolsky SA, Vetterling WT . Numerical Recipes in C: the Art of Scientific Computing . Chapter 11:pp 361 - 366, 1988, Jacobi Transformation . Cambridge University Press
Price TS, Regan R, Mott R, Hedman A, Honey B, Daniels RJ, Smith L, Greenfield A, Tiganescu A, Buckle V, Ventress N, Ayyub H, Salhan A, Pedraza-Diaz S, Broxholme J, Ragoussis J, Higgs DR, Flint J, Knight SJ . SW-ARRAY: a dynamic programming solution for the identification of copy-number changes in genomic DNA using array comparative genome hybridization data . Nucleic Acids Res 2005 ; 33 : 3455 - 3464
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, González JR, Gratacòs M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME . Global variation in copy number in the human genome . Nature 2006 ; 444 : 444 - 454
Rodriguez-Revenga L, Mila M, Rosenberg C, Lamb A, Lee C . Structural variation in the human genome: the impact of copy number variants on clinical diagnosis . Genet Med 2007 ; 9 : 600 - 606
Sasso EH, Buckner JH, Suzuki LA . Ethnic differences of polymorphism of an immunoglobulin VH3 gene . J Clin Invest 1995 ; 96 : 1591 - 1600
Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Månér S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M . Large-scale copy number polymorphism in the human genome . Science 2004 ; 305 : 525 - 528
Slater HR, Bailey DK, Ren H, Cao M, Bell K, Nasioulas S, Henke R, Choo KH, Kennedy GC . High-resolution identification of chromosomal abnormalities using oligonucleotide arrays containing 116,204 SNPs . Am J Hum Genet 2005 ; 77 : 709 - 726
Strachen T, Read AP . Human Molecular Genetics . 1999 ; Oxford: BIOS Scientific
Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, Olson MV, Eichler EE . Fine-scale structural variation of the human genome . Nat Genet 2005 ; 37 : 727 - 732
van der Burg M, Barendregt BH, van Gastel-Mol EJ, Tumkaya T, Langerak AW, van Dongen JM . Unraveling of the polymorphic Cλ2-Cλ3 amplification and the Ke+ Oz-polymorphism in the Human Igλ locus . J Immunol 2002 ; 169 : 271 - 276
Vaurs-Barriere C, Bonnet-Dupeyron MN, Combes P, Gauthier-Barichard F, Reveles XT, Schiffmann R, Bertini E, Rodriguez D, Vago P, Armour JA, Saugier-Veber P, Frebourg T, Leach RJ, Boespflug-Tanguy O . Golli-MBP copy number analysis by FISH, QMPSF and MAPH in 195 patients with hypomyelinating leukodystrophies . Ann Hum Genet 2006 ; 70 : 66 - 77
Wilson GM, Flibotte S, Chopra V, Melnyk BL, Honer WG, Holt RA . DNA copy-number analysis in bipolar disorder and schizophrenia reveals aberrations in genes involved in glutamate signaling . Hum Mol Genet 2006 ; 15 : 743 - 749
Yoo YK, Ke X, Hong S, Jang HY, Park K, Kim S, Ahn T, Lee YD, Song O, Rho NY, Lee MS, Lee YS, Kim J, Kim YJ, Yang JM, Song K, Kimm K, Weir B, Cardon LR, Lee JE, Hwang JJ . Fine-scale map of encyclopedia of DNA elements regions in the Korean population . Genetics 2006 ; 174 : 491 - 497
Acknowledgements
This work was supported by an intramural grant from Korea National Institute of Health, Korea Centers for Disease Control and Prevention (2007-N00353-00).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Jeon, JP., Shim, SM., Jung, J. et al. A comprehensive profile of DNA copy number variations in a Korean population: identification of copy number invariant regions among Koreans. Exp Mol Med 41, 618–628 (2009). https://doi.org/10.3858/emm.2009.41.9.068
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2009.41.9.068
