
Abstract
Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome is a genetically heterogeneous mitochondrial disorder with variable clinical symptoms. Here, from the sequencing of the entire mitochondrial genome, we report a Korean MELAS family harboring two homoplasmic missense mutations, which were reported 9957T > C (Phe251Leu) transition mutation in the cytochrome c oxidase subunit 3 (COX3) gene and a novel 13849A > C (Asn505His) transversion mutation in the NADH dehydrogenase subunit 5 (ND5) gene. Neither of these mutations was found in 205 normal controls. Both mutations were identified from the proband and his mother, but not his father. The patients showed cataract symptom in addition to MELAS phenotype. We believe that the 9957T > C mutation is pathogenic, however, the 13849A > C mutation is of unclear significance. It is likely that the 13849A > C mutation might function as the secondary mutation which increase the expressivity of overlapping phenotypes of MELAS and cataract. This study also demonstrates the importance of full sequencing of mtDNA for the molecular genetic understanding of mitochondrial disorders.
Similar content being viewed by others


Introduction
Several hundreds of different mitochondrial DNA (mtDNA) mutations have been reported with a variety of human disorders (http://mitomap.org). The mtDNA mutations are frequently divided into two groups, i.e., rearrangements and point mutations. Most rearrangements including large deletions are sporadic, however, point mutations are commonly inherited through the maternal lineage. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome (OMIM 540000) is one of the most frequent diseases caused by mtDNA point mutations. MELAS is a genetically heterogeneous mitochondrial disorder with variable clinical phenotypes, including focal or generalized seizures, recurrent headache, deafness, and diabetes (Pavlakis et al., 1984; Montagna et al., 1988).
Various pathogenic mtDNA mutations have been reported as the underlying causes of MELAS phenotype. In particular, more than 80% of MELAS cases have been reported to be associated with the tRNALeu (UUR) gene mutations including 3243A > G (Goto et al., 1990), 3251A > G (Sweeney et al., 1993), 3252T > C (Morten et al., 1993), and 3271T > C (Goto et al., 1991). For the mutations in polypeptide-coding genes associated with MELAS, 12770A > G resulting in Glu145Gly (Liolitsa et al., 2003), 13042G > A resulting in Ala236Thr (Naini et al., 2005), 13045A > C resulting in Met237Leu (Liolitsa et al., 2003), 13084A > T resulting in Ser250Cys (Crimi et al., 2003), and 13513G > A resulting in Asp393Asn (Santorelli et al., 1997) in the NADH dehydrogenase subunit 5 (ND5) gene and 9957T > C (Phe251Leu) in cytochrome c oxidase subunit 3 (COX3; Manfredi et al., 1995) have been reported. Recently, the ND5 has known to be a mutational hot spot for MELAS and other various overlapping diseases including myoclonus epilepsy with ragged-red fibers (MERRF), Leber's hereditary optic neuropathy (LHON) and Leigh syndrome (Liolitsa et al., 2003; Naini et al., 2005; Valentino et al., 2006). The A11084G (Thr109Ala) in the ND4 gene was also reported to be associated with MELAS (Lertrit et al., 1992). However, it was later suggested to be a polymorphism (Sakuta et al., 1993). MtDNA point mutations found in MELAS patients are usually present as the heteroplasmic condition rather than homoplasmy.
Since several pathogenic mutations of LHON have been preferentially associated with haplogroup J (Brown et al., 1997; Hofmann et al., 1997; Torroni et al., 1997; Carelli et al., 2006), the association studies between haplogroups and diseases have became an important approach to uncover the function of mtDNA variations. However, no finding was still reported that MELAS is preferentially associated with any specific haplogroup.
In the present study, we examined a Korean family with various MELAS symptoms. In addition, this family revealed the clinical phenotype of cataract. From the sequencing of the entire mitochondrial genomes from the family samples, we identified two missense mutations, 9957T > C (Phe251Leu) in the COX3 gene and 13849A > C (Asn505His) in the ND5 gene. The 9957T > C in COX3 has been previously reported in MELAS and non-arteritic anterior ischemic optic neuropathy (NAION) patients; however, the 13849A > C transversion mutation in ND5 was determined to be novel.
Materials and Methods
Subjects and DNA extraction
This study involved three members (affecteds: proband and his mother, unaffected: his father) from a family with MELAS and cataract phenotypes (familial ID: MT48, Figure 1A). A group of 205 non-MELAS healthy controls were also involved for this study. Total DNA was extracted from whole blood using a QIAamp DNA blood mini kit (Qiagen, Germany). The paternity and maternity of the MT48 family were confirmed by genotyping of 15 short tandem repeat (STR) markers using a PowerPlex 16 system (Promega, Madison, WI). All participants provided written informed consent according to the protocol approved by the Ethics Committee of Ewha Womans University Hospital (Seoul, Korea).

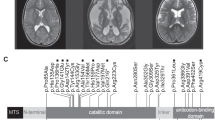
Pedigree of a MELAS family and diffusion-weighted imaging of the patient's brain. (A) Pedigree of family with MELAS and cataract phenotypes (familial ID:MT48). Black symbols indicate affected members, and open symbol indicates unaffected member. (B) Diffusion-weighted imaging of the patient 1's brain (II-1). The MRI feature demonstrated acute cortical infarct, and revealed a right frontal (arrow) and temporal (arrowhead) hyperintense signals mainly involving the gray matter.
Sequencing analysis of whole mitochondrial genome
The entire mitochondrial genome was amplified by PCR as 46 overlapping fragments using a MitoSEQr resequencing system (Applied Biosystems, Foster City, CA). The PCR amplification condition consisted of initial denaturation at 96℃ for 5 min, followed by 35 cycles at 94℃ for 30 s, 60℃ for 45 s, 72℃ for 45 s, and a final extension at 72℃ for 10 min. PCR products were purified by a EXOSAP-IT kit (USB, USA) and nucleotide sequence was determined by an automatic genetic analyzer ABI3100 using the big dye terminator cycle sequencing ready reaction kit (Applied Biosystems). Sequence variations were confirmed by analyzing both strands of DNA. The obtained sequences were compared with the revised Cambridge reference sequence (rCRS) (Anderson et al., 1981; Andrews et al., 1999) using SeqScape software (Applied Biosystems). Haplogrouping of mutations from the entire mtDNA sequence was followed by Kong et al. (2003, 2006) and Salas et al. (2005).
Determination of heteroplasmy
Heteroplasmy of the mtDNA mutations were determined by sequencing plasmid clones having the corresponding mtDNA fragment. The DNA fragments including the mutation site were obtained by PCR amplification, and were subcloned into the pGEM-T easy vector (Promega), which was then used to transform E. coli. Plasmid DNA was isolated from 100 random picked colonies per a patient using a plasmid DNA isolation kit (SolGent, Korea), and the mutation was determined by the sequencing of the insert DNA. The proportion of heteroplasmy was measured by the counting of clones with mutant mtDNA from total 100 clones.
Clinical assessment and MRI study
Clinical information was obtained from a standardized manner. The clinical examination included assessments of cranial nerves, motor and sensory impairments, and muscle atrophy. Eye examinations included fundus, ophthalmoscopy, color vision using Ishihara color plates, visual evoked potential (VEP), and brainstem auditory evoked potential (BAEP). Motor and sensory conduction velocities of median, ulnar, peroneal, tibial, and sural nerves were determined. A muscle biopsy was taken from the quadriceps under local anesthesia. Cross-sections of biopsy tissue were stained with hematoxylin-eosin, modified Gomori trichrome, cytochrome c oxidase, and succinate dehydrogenase. Another biopsy fragment was examined by electron microscopy. MRI was performed in a supine position using a 1.5-T system (Siemens Vision; Siemens, Erlangen, Germany). Whole brains were scanned using a slice thickness of 7 mm and a 2-mm interslice gap to produce 16 axial images.
Results
Clinical phenotypes of patients
Patient 1 (proband): A 25-year-old male (II-1 in Figure 1A) was admitted due to a sudden development of weakness on the left side and generalized seizure. At 5 years of age, he was diagnosed as diabetes and hypothyroidism. He had a short stature with a height of 153 cm, and had experienced recurrent migraine without aura. Ophthalmological examination showed bilateral cataracts, but no diabetic retinopathy and NAION. The VEP and BAEP were normal. Ptosis, ophthaloplegia, and hearing loss were not observed. Cardiological examinations revealed no evidence of an embolic event. Electromyography showed mild myopathic changes, and nerve conduction studies were normal. Laboratory examinations revealed elevated lactate (29.0; normal: 4.5-19.8 mg/dl), and hemoglobin A1C (HbA1C) level (8.6; normal: 4-6%). Thyroid stimulating hormone (TSH) was elevated (9.17; normal: 0.3-5.0 µIU/ml), but triiodothyronine (T3) (42.4; normal: 80-200 mg/dl) and thyroxine (T4) (4.4; normal: 4.5-12.5 mg/dl) were decreased. Diffusion-weighted brain MRI showed acute cerebral infarction, and a high signal intensity area in right frontal and temporal lobes, mainly involving the gray matter (Figure 1B). In addition, some cortical and cerebellar atrophy was found. Analysis of a quadriceps muscle biopsy specimen showed a few ragged-red fibers, which was consistent with the findings of a previous case with the 9957T > C mutation (Manfredi et al., 1995). Semi-thin transverse sections showed multiple megaconia in the intermyofibrillar area and the subsarcolemmal area (data not shown).
Patient 2: The mother of patient 1 is a 54-year-old female (I-2 in Figure 1A). She also had a short stature (154 cm), and experienced recurrent migraine. She was diagnosed as rheumatoid arthritis at 39 years. Bilateral cataracts were found at age 27; however, ocular and lid movements were normal. At 45 years of age, she underwent successful bilateral cataract extraction with intraocular lens implantation. Laboratory examination displayed elevated lactate level (21.2 mg/dl). Her brain MRI showed bilateral non-specific periventricular hyperintnsities.
Identification of mtDNA mutations
From the sequencing analysis of the whole mtDNA in MELAS patients from MT48 family, several decades of variants were identified, compared with rCRS. The phylogenetic analysis suggested the most mutations in the patient's mitochondrial genome seems to be a M7c2 haplogroup-specific (Figure 3). The (sub)haplogroup M is one of the prevalent haplogroups in southeast Asian ethnic population, and is also frequent in northern Asia (Kivisild et al., 2002; Kong et al., 2003, 2006). Table 1 showed entire variants observed in the patients except for variants in control region. Of them, we were able to identify two missense mutations as the suspected underlying causes of MELAS, the 9957T > C transition mutation resulting in Phe251Leu in the COX3 gene and the 13849A > C transversion mutation resulting in Asn505His in the ND5 gene (Figure 2A). These two mutations were found in the proband (DNA from blood and muscle) and his mother (DNA from blood), but were not found in his unaffected father. Both mutations were determined to be a homoplasmic states. These two mutations were not found in the 205 normal controls.

Haplogroup classification based on complete mtDNA genome sequence in the MT48 patients. The mtDNA mutations identified in the patient suggested M7c2 haplogroup-specific. Numbers refer to substitutions at nucleotide positions with respect to the revised Cambridge reference sequence (rCRC). The suffixes indicate transversions, and underlines indicate recurrent mutations. Others indicate transitions. Open boxes represent mutations observed in the patients implicating the M7c2 haplogroup, and grayed box represents the private mutations specific for MT48 family.

Identification of two causative missense mutations in MT48 MELAS family. (A) Sequence chromatograms of the 9957T > C (Phe251Leu) mutation in CO3 (left) and the 13849A > C (Asn505His) mutation in ND5 (right). (B) Conservation of these sequences in different species. The 9957T > C (Phe251Leu) mutation site is highly conserved and the 13849A > C (Asn505His) mutation is moderately conserved.
The 13849A > C (Asn505His) mutation in ND5 has not been previously reported in the MITOMAP database (http://www.mitomap.org). The 9957T > C (Phe251Leu) mutation in the COX3 gene was first reported in a MELAS patient (Manfredi et al., 1995), but thereafter, an association with NAION was also suggested by Abu-Amero et al. (2005). The site of the 9957T > C (Phe251Leu) mutation was highly conserved in different species, while the 13849A > C mutation was moderately conserved in different species (Figure 2B). No causative mutation was found in the tRNALeu (UUR) gene, which provides most frequently genetic defects of MELAS phenotype.
We also found many polymorphic variants through whole mtDNA sequencing analysis. The reported missense variants in the coding regions are 5442T > C (Phe > Leu) in ND2, 8701A > G (Thr > Ala) and 8860A > G (Thr > Ala) in ATP6, 10398A > G (Thr > Ala) and 10400C > T (Thr > Ala) in CO3, 14180T > C (Thr > Cys) in ND6, and 15326A > G (Thr > Ala) in CytB. We also identified two synonymous variants in the coding regions, 11665C > T and 12091T > C in ND4, which were not reported in the MITOMAP database (http://www.mitomap.org), but involved in M7c-specific haplogroup. We did not consider these two mutations as pathogenic because they did not change amino acids and were also found in control samples (rare allele frequencies of 11665C > T and 12091T > C: 0.047 and 0.043, respectively).
Discussion
From the whole mitochondrial genome analysis, we identified two missense mutations, the 9957T > C (Phe251Leu) in COX3 and the 13849A > C (Asn505His) in ND5 as the homoplasmic states (Figure 2A). The 9957T > C mutation was previously reported to show an association of MELAS with neurologic symptoms such as stroke, seizure, and lactic acidosis (Manfredi et al., 1995). Thereafter, it was also found in a NAION patient (Abu-Amero et al., 2005). Our case showed no NAION phenotype, whereas, he revealed typical MELAS symptom with additional phenotype of bilateral cataracts.
Several ND5 mutations associated with MELAS have been recently reported (Santorelli et al., 1997; Crimi et al., 2003; Naini et al., 2005). It is now recognized that mutations in ND5 are the second most frequent genetic causes of MELAS following the tRNALeu (UUR) gene. Particularly, mutations in ND5 gene have frequently revealed various overlapping phenotypes including MELAS, LHON, MERRF and Leigh syndrome (Liolitsa et al., 2003; Naini et al., 2005; Valentino et al., 2006). The heteroplasmic 13042G > A mutation was found in patient with overlap syndromes including MELAS and stroke-like MERRF (Naini et al., 2005) and patient with a prevalent ocular phenotype including LHON-like optic neuropathy, retinopathy, cataract, strokes, early deaths, and miscarriages (Valentino et al., 2006). The 13045A > C mutation showed phenotypes of MELAS, LHON and Leigh's overlap syndrome (Liolitsa et al., 2003). The novel 13849A > C (Asn505His) mutation in this study is also located in ND5.
Cataract, the opacification of the eye lens, is the leading cause of blindness worldwide. It has been reported that opacity of the lens is a direct result of oxidative stress, and mitochondria is a key organelle in the production of oxidative stress. Juvenile cataract in patients with mitochondrial disease is well documented (Isashiki et al., 1998). Even among patients with mitochondrial disease, there could be a subset of patients with a different susceptibility to cataract formation. Even though patient 1 was diagnosed with diabetes, his mother (patient 2) did not have diabetes, which indicates that cataract probably was not associated with diabetes in this family. The cataract symptom in the MT48 family might be associated with the 13849A > C (Asn505His) mutation.
We regarded that the 9957T > C (Phe251Leu) mutation in COX3 is an underlying cause of MELAS phenotype of MT48 family as the following reasons; (1) co-segregation of the mutation with affected members in the pedigree, (2) no detection of same mutation in 205 control samples, (3) well conservation of amino acid at the mutation site among different species, and (4) phenotypic similarity with the previous case with 9957T > C mutation (Manfredi et al., 1995). However, the 13849A > C mutation in ND5 is of unclear functional significance, although it was not found in 205 controls and also not reported as any haplogroup-specific mutation. The mutation was found as homoplasmic in compared that general pathogenic mutations in mtDNA are commonly heteroplasmic (Figure 2A), and the mutation site was not highly conserved among different species (Figure 2B), thus, it may be the 13849A > C private mutation is just a rare polymorphism.
Recently, several secondary mutations have been reported in LHON and MELAS patients (Torroni et al., 1997; Tzen et al., 2003; Li et al., 2006; Qu et al., 2006). It is likely that the 13849A > C mutation might function as the secondary mutation increasing the expressivity of overlapped phenotypes of MELAS and cataract. However, further studies such as investigation of different ethnic groups and biochemical enzyme activity in muscle biopsy should be performed to confirm the 13849A > C mutation as pathogenic.
Abbreviations
- COX3:
-
cytochrome c oxidase subunit 3
- MELAS:
-
mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes
- MERRF:
-
myoclonus epilepsy with ragged-red fibers
- mtDNA:
-
mitochondrial DNA
- NAION:
-
non-arteritic anterior ischemic optic neuropathy
- ND5:
-
NADH dehydrogenase subunit 5
- rCRS:
-
revised Cambridge reference sequence
References
Abu-Amero KK, Bosley TM, Bohlega S, Hansen E . Mitochondrial T9957C mutation in association with NAION and seizures but not MELAS . Ophthalmic Genet 2005 ; 26 : 31 - 36
Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG . Sequence and organization of the human mitochondrial genome . Nature 1981 ; 290 : 457 - 465
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N . Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA . Nat Genet 1999 ; 23 : 147 -
Brown MD, Sun F, Wallace DC . Clustering of Caucasian Leber hereditary optic neuropathy patients containing the 11778 or 14484 mutations on an mtDNA lineage . Am J Hum Genet 1997 ; 60 : 381 387 -
Carelli V, Achilli A, Valentino ML, Rengo C, Semino O, Pala M, Olivieri A, Mattiazzi M, Pallotti F, Carrara F, Zeviani M, Leuzzi V, Carducci C, Valle G, Simionati B, Mendieta L, Salomao S, Belfort R, Sadun AA, Torroni A . Haplogroup effects and recombination of mitochondrial DNA: novel clues from the analysis of Leber hereditary optic neuropathy pedigrees . Am J Hum Genet 2006 ; 78 : 564 - 574
Crimi M, Galbiati S, Moroni I, Bordoni A, Perini MP, Lamantea E, Sciacco M, Zeviani M, Biunno I, Moggio M, Scarlato G, Comi GP . A missense mutation in the mitochondrial ND5 gene associated with a Leigh-MELAS overlap syndrome . Neurology 2003 ; 60 : 1857 - 1861
Goto Y, Nonaka I, Horai S . A mutation in the tRNALeu (UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies . Nature 1990 ; 348 : 651 - 653
Goto Y, Nonaka I, Horai S . A new mtDNA mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episode (MELAS) . Biochim Biophys Acta 1991 ; 1097 : 238 - 240
Hofmann S, Jaksch M, Bezold R, Mertens S, Aholt S, Paprotta A, Gerbitz KD . Population genetics and disease susceptibility: characterization of central European haplogroups by mtDNA gene mutations, correlation with D loop variants and association with disease . Hum Mol Genet 1997 ; 6 : 1835 - 1846
Isashiki Y, Nakagawa M, Ohba N, Kamimura K, Sakoda Y, Higuchi I, Izumo S, Osame M . Retinal manifestations in mitochondrial diseases associated with mitochondrial DNA mutation . Acta Ophthalmol Scand 1998 ; 76 : 6 - 13
Kivisild T, Tolk HV, cParik J, Wang Y, Papiha SS, Bandelt HJ, Villems R . The emerging limbs and twigs of the East Asian mtDNA tree . Mol Biol Evol 2002 ; 19 : 1737 - 1751
Kong QP, Yao YG, Liu M, Shen SP, Chen C, Zhu CL, Palanichamy MG, Zhang YP . Mitochondrial DNA sequence polymorphisms of five ethnic populations from northern China . Hum Genet 2003 ; 113 : 391 - 405
Kong QP, Bandelt HJ, Sun C, Yao YG, Salas A, Achilli A, Wang CY, Zhong L, Zhu CL, Wu SF, Torroni A, Zhang YP . Updating the East Asian mtDNA phylogeny: a prerequisite for the identification of pathogenic mutations . Hum Mol Genet 2006 ; 15 : 2076 - 2086
Lertrit P, Noer AS, Jean-Francois MJ, Kapsa R, Dennett X, Thyagarajan D, Lethlean K, Byrne E, Marzuki S . A new disease-related mutation for mitochondrial encephalopathy lactic acidosis and stroke-like episodes (MELAS) syndrome affects the ND4 subunit of the respiratory complex I . Am J Hum Genet 1992 ; 51 : 457 - 468
Li R, Qu J, Zhou X, Tong Y, Hu Y, Qian Y, Lu F, Mo JQ, West CE, Guan MX . The mitochondrial tRNA(Thr) A15951G mutation may influence the phenotypic expression of the LHON-associated ND4 G11778A mutation in a Chinese family . Gene 2006 ; 376 : 79 - 86
Liolitsa D, Rahman S, Benton S, Carr LJ, Hanna MG . Is the mitochondrial complex I ND5 gene a hot-spot for MELAS causing mutations ? Ann Neurol 2003 ; 53 : 128 - 132
Manfredi G, Schon EA, Moraes CT, Bonilla E, Berry GT, Sladky JT, DiMauro S . A new mutation associated with MELAS is located in a mitochondrial DNA polypeptide-coding gene . Neuromuscul Disord 1995 ; 5 : 391 - 398
Montagna P, Gallassi R, Medori R, Govoni E, Zeviani M, DiMauro S, Lugaresi E, Andermann F . MELAS syndrome: characteristic migrainous and epileptic features and maternal transmission . Neurology 1988 ; 38 : 751 - 754
Morten KJ, Cooper JM, Brown GK, Lake BD, Pike D, Poulton J . A new point mutation associated with mitochondrial encephalomyopathy . Hum Mol Genet 1993 ; 2 : 2081 - 2087
Naini AB, Lu J, Kaufmann P, Bernstein RA, Mancuso M, Bonilla E, Hirano M, DiMauro S . Novel mitochondrial DNA ND5 mutation in a patient with clinical features of MELAS and MERRF . Arch Neurol 2005 ; 62 : 473 - 476
Pavlakis SG, Phillips PC, DiMauro S, DeVivo DC, Rowland LP . Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes: a distinctive clinical syndrome . Ann Neurol 1984 ; 16 : 481 - 488
Qu J, Li R, Zhou X, Tong Y, Lu F, Qian Y, Hu Y, Mo JQ, West CE, Guan MX . The novel A4435G mutation in the mitochondrial tRNAMet may modulate the phenotypic expression of the LHON-associated ND4 G11778A mutation . Invest Ophthalmol Vis Sci 2006 ; 47 : 475 - 483
Sakuta R, Goto YI, Nonaka I, Horai S . An A-to-G transition at nucleotide pair 11084 in the ND4 gene may be an mtDNA polymorphism . Am J Hum Genet 1993 ; 53 : 964 - 965
Salas A, Yao YG, Macaulay V, Vega A, Carracedo A, Bandelt HJ . A critical reassessment of the role of mitochondria in tumorigenesis . PLoS Med 2005 ; 2 : e296 -
Santorelli FM, Tanji K, Kulikova R, Shanske S, Vilarinho L, Hays AP, DiMauro S . Identification of a novel mutation in the mtDNA ND5 gene associated with MELAS . Biochem Biophys Res Commun 1997 ; 238 : 326 - 328
Sweeney MG, Bundey S, Brockington M, Poulton JR, Winer JB, Harding AE . Mitochondrial myopathy associated with sudden death in young adults and a novel mutation in the mitochondrial DNA transfer RNA[Leu(UUR)] gene . Q J Med 1993 ; 86 : 709 - 713
Torroni A, Petrozzi M, D'Urbano L, Sellitto D, Zeviani M, Carrara F, Carducci C, Leuzzi V, Carelli V, Barboni P, De Negri A, Scozzari R . Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484 . Am J Hum Genet 1997 ; 60 : 1107 - 1121
Tzen CY, Thajeb P, Wu TY, Chen SC . MELAS with point mutations involving tRNALeu (A3243G) and tRNAGlu (A14693G) . Muscle Nerve 2003 ; 28 : 575 - 581
Valentino ML, Barboni P, Rengo C, Achilli A, Torroni A, Lodi R, Tonon C, Barbiroli B, Fortuna F, Montagna P, Baruzzi A, Carelli V . The 13042G→A/ND5 mutation in mtDNA is pathogenic and can be associated also with a prevalent ocular phenotype . J Med Genet 2006 ; 43 : e38 -
Acknowledgements
This work was supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD:KRF-2005-041-F00025), and the star project in Kongju National University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Choi, BO., Hwang, J., Kim, J. et al. A MELAS syndrome family harboring two mutations in mitochondrial genome. Exp Mol Med 40, 354–360 (2008). https://doi.org/10.3858/emm.2008.40.3.354
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2008.40.3.354
Keywords
This article is cited by
-
Next-generation sequencing profiling of mitochondrial genomes in gout
Arthritis Research & Therapy (2018)
-
The identification of mitochondrial DNA variants in glioblastoma multiforme
Acta Neuropathologica Communications (2014)
-
Populationsgenetische mitochondriale DNA-Daten
Medizinische Genetik (2008)
