
Abstract
Variants in the TTN gene have been associated with distal myopathies and other distinctive phenotypes involving skeletal and cardiac muscle. Through whole-exome sequencing we identified a novel stop-gain variant (c.107635C>T, p.(Gln35879Ter)) in the TTN gene, coding a part of the M-line of titin, in 14 patients with autosomal recessive distal myopathy and Serbian ancestry. All patients share a common 1 Mb core haplotype associated with c.107635C>T, suggesting a founder variant. In compound heterozygotes, nine other TTN variants were identified: four stop-gain, three frameshift, one missense and one splice donor variant. Patients homozygous for the common variant did not show significant clinical differences to the compound heterozygous patients. The clinical presentation of all patients was an adult onset distal myopathy with predominant lower limb involvement. In addition, most patients had normal to mildly elevated serum creatine kinase levels, myopathic electromyograms, normal cardiologic and respiratory tests and muscle pathology consistent with a dystrophic process. In this study, we describe a distinct phenotype for patients with distal myopathy associated with novel recessive TTN variants including a Serbian founder variant. Our results expand the phenotypic and genetic spectrum of titinopathies and will facilitate the diagnosis of this condition in patients of Serbian origin.
Similar content being viewed by others


Introduction
Distal myopathies are a group of progressive hereditary muscle disorders characterised by the onset of weakness and wasting in the lower legs, forearms, feet and/or hands.1 They are less frequent and less well studied than many muscular dystrophies affecting predominantly proximal muscles and therefore probably often underdiagnosed. There is a growing number of distinctive and genetically defined distal myopathies with either autosomal dominant or recessive inheritance. It has been speculated that the selectivity of distal or proximal muscle involvement could be related to the subcellular localisation of the defective proteins: sarcomeric defects generally cause a more distal pattern of involvement, while sarcolemmal defects are more frequently responsible for proximal phenotypes.2
The TTN gene (OMIM 188840), located on chromosome 2q31, consists of 363 coding exons. It encodes titin, the largest protein known in humans. After myosin and actin, titin is the third most common filament in both skeletal and cardiac muscle. The molecular structure of titin is divided into four regions: the N-terminal Z-disc, the I-band and A-band regions and the C-terminal M-line, encoded by the last six exons of the TTN gene (358–363, or Mex1 to Mex6). Disorders caused by variants in the TTN gene can be classified into three groups: cardiomyopathies, diseases of skeletal muscles and congenital disorders affecting both types of muscles.3 Before the application of next-generation sequencing technologies, it was very difficult to diagnose conditions associated with TTN variants because of the size of the gene.4, 5 Even now it is difficult to determine the pathological variants because of the complex and repetitive gene structure and the fact that almost every individual carries at least one rare, mostly non-pathogenic TTN variant.
Several distinctive phenotypes involving skeletal muscles have been associated with variants in the TTN gene. Tibial muscular dystrophy (TMD; OMIM 600334) is a late-onset, autosomal dominant disorder characterised by weakness and wasting predominantly in the anterior compartment of the distal lower limbs.1 It was first described in Finland where it represents the most common muscle disease.6 The FINmaj variant is the most prevalent TTN variant in the Finnish population (~2:10 000) due to a founder effect. It is a complex 11-bp insertion/deletion resulting in a 4-amino-acid exchange in the last exon (Mex6, exon 363) of the TTN gene.7 Several other variants localised in the second to last (Mex5, exon 362) and the last exons of TTN have been described in TMD patients from Belgian, French, Spanish and Italian populations.8, 9, 10, 11 Homozygosity of the FINmaj variant is rare and causes limb-girdle muscular dystrophy 2J (OMIM 608807), a phenotype with earlier onset and more proximal muscle involvement.1, 6, 10 Recently, atypical, more complex or severe phenotypes have been explained by a second TTN variant in several TMD patients from different European countries and in LGMD2J patients being compound heterozygous for the FINmaj variant and an additional TTN variant.12 Hereditary myopathy with early respiratory failure (OMIM 603689), an autosomal dominant disease involving proximal, distal and respiratory muscles, represents yet another entity caused by TTN variants.13 The spectrum of titinopathies is constantly being broadened.
In the present study, we identified recessive TTN variants as a molecular cause of distal myopathy resembling TMD in a well-defined cohort of patients with Serbian ancestry through whole-exome sequencing (WES). A stop-gain TTN variant (c.107635C>T, p.(Gln35879Ter)) was found in all patients (hereafter referred as the ‘common variant’) – 3 patients being homozygotes and 11 compound heterozygotes. Additionally, we showed that the common variant was part of a haplotype block that likely originated from a single founder event. Our findings suggest that screening for the identified founder TTN variant should be the first step in the diagnostic algorithm of distal myopathies in patients with Serbian ancestry.
Materials and methods
Clinical assessment
Patients were recruited at the Neurology Clinic, Clinical Centre of Serbia, School of Medicine, University of Belgrade and the Clinic for Neurology and Psychiatry for Children and Youth, Belgrade, Serbia as a part of the international MYO-SEQ project. This project provides WES for 1000 index patients with unexplained limb-girdle muscle weakness and elevated serum CK levels. Initial clinical details were submitted through PhenoTips,14 a tool developed for sharing phenotypic and genetic data in a safe and ethical way using the Human Phenotype Ontology.15 Appropriate informed consent was provided by all the patients after the Ethics Committee of the School of Medicine, University of Belgrade approved this study – no. 29/VI-18. The initial analysis included 91 unrelated index patients, with 19 having a predominantly distal phenotype. In this group with a distal phenotype, 14 patients from 14 unrelated Serbian families carried a specific TTN founder variant and were analysed further in this study. Among the five remaining patients with distal phenotype, three have been diagnosed with dysferlinopathy, one with desminopathy, and one remained without a conclusive genetic diagnosis.
The majority of patients (12/14) were sporadic, while two pedigrees suggested autosomal recessive inheritance. Consanguinity was probably present in one family (patient no. 02). First-degree relatives of patients were also assessed where possible. Clinical assessment included a detailed medical and family history and neurological examination, performed by an experienced neurologist. Muscle strength was scored using the Medical Research Council Muscle Grading Scale.16 Follow-up visits were regularly scheduled for all patients. Electromyography (EMG) and nerve conduction studies were performed in all patients. Serum CK levels were measured in all patients, and where possible in available family members. Respiratory function was assessed by spirometry annually, while regular cardiac examinations included ECG each year and echocardiogram every second year. Muscle biopsies were performed in seven patients.
Muscle magnetic resonance imaging (MRI) was performed in six patients in axial and coronal planes of the lower limbs using the following sequences: T1-weighted (T1w), T2-weighted, proton-density weighted and three-point Dixon.17, 18 Images were assessed on an individual muscle basis and graded according to the five-point scale published by Mercuri et al.19
Whole-exome sequencing
DNA was extracted from blood using standard techniques at the Neurology Clinic, Clinical Centre of Serbia, School of Medicine, University of Belgrade, Serbia. WES was performed at the Broad Institute of Harvard and MIT (Cambridge, MA, USA), using Illumina exome capture, 38 Mb baited target, and their in-solution hybrid selection process with >250 ng of input DNA (at >2 ng/μl). The sequencing pipeline included sample plating, library preparation (2-plexing of samples per hybridisation), hybrid capture, sequencing (76 bp paired reads), sample identification quality control check and data storage. The hybrid selection libraries cover >80% of targets at 20x, with a mean target coverage of >80x. The exome sequencing data was demultiplexed and each sample’s sequence data was aggregated into a single Picard BAM file. Alignment was carried out against the human reference hg19, build 37 using Burrows-Wheeler Aligner and variants were called using the Genome Analysis Toolkit software (Broad Institute, Cambridge, MA, USA). Data were analysed using the Broad Institute open source platform seqr (https://seqr.broadinstitute.org). The selection of potential variants from the whole-exome analysis was restricted to 169 genes that are known to be associated with neuromuscular disorders. Filtering criteria of potential variants included population frequency of <1% and moderate to high impact on protein structure (ie, missense, stop-gain, splice site, frameshift and in-frame indels). TTN variants and exons were annotated according to transcript ENST00000589042.1 and protein sequence ENSP00000467141.1 (RefSeq NM_001267550 and NP_001254479).
Validation of identified genetic variants
Sanger sequencing was used to confirm the presence of variants identified by WES in 13 patients (DNA sample from patient no. 04 was not available), and to perform segregation analysis in eight families, involving 18 first-degree relatives. Sanger sequencing was carried out with the BigDye Terminator v.1.1 Cycle Sequencing Kit (Life Technologies, Grand Island, NY, USA) on the Applied Biosystems 3130 Genetic Analyser (Applied Biosystems, Warrington, UK) at the Centre for Human Molecular Genetics, Faculty of Biology, University of Belgrade, Serbia. Additionally, 103 control individuals of Serbian origin were screened for the common c.107635C>T variant at the John Walton Muscular Dystrophy Research Centre, Institute of Genetic Medicine, Newcastle University, UK. All identified variants were submitted to the European Genome-Phenome Archive (https://www.ebi.ac.uk/) and the TTN Leiden Open Variation Database (http://www.lovd.nl/TTN; patient IDs 87088, 87240–87245 and 87247– 87255).
Haplotype analysis
The size of the haplotype associated with the common c.107635C>T variant was first estimated using WES data for the homozygous patients and a homozygosity block was identified by running the full Variant Caller Format files on Homozygosity Mapper (http://www.homozygositymapper.org/). In addition, an Illumina Infinium array containing ~250 000 markers allowed the identification of homozygosity blocks based on non-coding single-nucleotide polymorphisms (SNPs). From this, a 2.8 Mb window surrounding the TTN gene, containing 230 SNPs with an allele frequency >1%, was analysed in all patients. As no parental data were available, haplotypes of compound heterozygous cases were phased by inferring the genotype based on the common founder haplotype observed in the homozygous cases. The haplotype segregating with the c.107635C>T variant was also reconstructed by genotyping analyses of eleven polymorphic microsatellites located upstream (D2S1776, D2S2188, D2S2314, D2S138, D2S148 and D2S300), within (D2S385) and downstream (D2S384, D2S2310, D2S364 and D2S152) of the TTN gene. Microsatellite haplotypes were phased by family segregation analysis, except for patient nos 05, 08 and 13 in whom the haplotypes were phased as described above for SNPs haplotypes. Genotyping was performed by fragment analysis on Applied Biosystems 3130 Genetic Analyser (Applied Biosystems) and data were analysed with GeneMapper ID v.3.2.1 software (Applied Biosystems).
The age of the most recent common ancestor of c.107635C>T variant in the analysed sample ('mutation age') was estimated by the single locus method based on the expected exponential decay of linkage disequilibrium between the variant and nearby polymorphic loci (D2S314, D2S138, D2S148, D2S2310 and D2S364) through recombination over time.20, 21 Microsatellite alleles segregating with the variant and their frequencies were determined from haplotype data of 14 disease chromosomes allowing ±1 repeat unit, while allele frequencies in the Serbian population were obtained from 56 normal chromosomes. To estimate the recombination fraction (Θ) between a given marker and the variant, physical distances were converted into genetic distances using the deCODE sex-averaged recombination rates.
Results
Recessive TTN variants are a frequent cause of distal myopathy in Serbia
Through WES we identified a novel stop-gain variant (c.107635C>T, p.(Gln35879Ter)) in 14 patients (out of 19) with distal myopathy of Serbian ancestry. This variant is located in exon 362 (Mex 5), coding a part of the M-line region of titin. Three patients (nos 02, 05 and 13) were homozygous for the variant (Table 1). Two heterozygous carriers of European (non-Finnish) origin are reported in the Exome Aggregation Consortium (ExAC) data set. As the Serbian population might not be well represented among the >60 000 individuals of ExAC, we screened a group of 103 control individuals of Serbian origin. The common variant was absent, suggesting that it is not a frequent benign variant in the Serbian population.
In all 11 patients heterozygous for the common variant, a second variant in the TTN gene was detected (Table 1). Three of those patients (nos 01, 08 and 14) had the same frameshift variant c.103360delG; p.(Glu34454AsnfsTer3), located in Mex1 (exon 358). Only two additional frameshift variants, four stop-gain, one missense and one splice donor variant, were found in the rest of the patients. Among those variants, two were located in Mex1, one was in the TTN exons encoding the I-band regions of titin and others were in exons encoding the A-band (Table 1). The missense variant is found in ExAC (MAF=8.3 × 10−6, allele count is 1 out of 120 160).
Sanger sequencing confirmed the presence of the WES-identified TTN variants in 13 patients (DNA sample of one patient was not available). According to the family segregation analysis, all parents were heterozygous carriers for one of the TTN variants identified in patients, and among other tested first-degree relatives, six out of seven were heterozygous carriers (Supplementary Table 1). All relatives who were heterozygous carriers of those variants were asymptomatic, which is in accordance with the presumed autosomal recessive mode of inheritance (Figure 1).

Segregation analysis of TTN variants in the family of patient no. 01. Both unaffected parents carry one of the variants identified in the patient, which is in accordance with an autosomal recessive mode of inheritance. The unaffected brother is a carrier of the wild-type alleles. The pedigree symbol for the patient is shown in black.
Two identified TTN variants are founder variants
Initial homozygosity analysis based on exonic variants of patient nos 02 and 05, both homozygous for the common variant, indicated that the size of a common haplotype associated with the c.107635C>T variant was ~4.8 Mb. Further detailed analysis of genome-wide SNP array data, including non-coding SNPs, delineated the size of the core haplotype (the part of the ancestral haplotype shared by all c.107635C>T chromosomes) to just over 1 Mb between rs334613:A>G (hg19 chr2:g.179166242A>G) and rs7591863:A>G (hg19 chr2:g.180196356A>G). The common haplotype was also confirmed by fine mapping of polymorphic microsatellites and some of them were in strong linkage disequilibrium with the common variant (Supplementary Table 2). Taking all these data together the size of the core haplotype was estimated to be 1 Mb (Figure 2). In addition, microsatellite analysis also showed a common haplotype of at least 7.4 Mb for the c.103360delG variant, which was identified in three patients (data not shown). These results support a common origin for both variants, c.107635C>T and c.103360delG, in Serbian patients, with c.107635C>T possibly representing a major Serbian TTNfounder variant.

Identification of the core founder haplotype. Haplotypes segregating with the common c.107635C>T variant defined by genome-wide SNP array data and microsatellite markers. All analysed chromosomes carrying the c.107635C>T variant (in orange) share the 1 Mb core haplotype, between rs334613 and rs7591863 (in grey). SNPs indicating a breakpoint are shown in red. Genotypes that could not be fully phased are shown in italics. Genomic locations are from the Human Genome Assembly GRCh37. Microsatellite allele size is shown in bp. First, last and every tenth SNP are shown.
The majority of c.107635C>T chromosomes shared a haplotype that exceeded the size of the core haplotype, indicating that recombination events shaped the ancestral haplotype in the past and thus enabling the estimation of the 'mutation age'. The mean age of the most recent common ancestor of the c.107635C>T variant in our sample was estimated at 40.9±11.2 generations (95% CI: 29.7–52.1). This corresponds to 820±223 (95% CI: 625–1015) or 1023±280 (95% CI: 778–1268) years assuming an average rate of 20 and 25 years per generation, respectively (Supplementary Table 2).
Patients with the common c.107635C>T TTN variant have a homogenous phenotype
We analysed phenotypic data of the 14 patients harbouring the c.107635C>T TTN variant in our cohort. Eight patients were female (57%) and six were male (43%) with an average age at the time of the last examination of 36.8±10.9 years. The mean disease duration was 7.8±6.1 years, not taking into account patient no. 04, for whom the age of onset was unknown. The clinical presentation associated with the common c.107635C>T variant was distal myopathy resembling TMD.
The age of onset ranged between 14 and 44 years (mean 28.0±9.4 years, median 25 years; Tables 2 and 3). The youngest patient (no. 14) was oligosymptomatic, with mild hyperCKemia, Achilles tendon (AT) contractures and a tendency of toe walking as the only clinical signs. Since the age of 9 years, patient no. 14 complained of calf pain following extensive physical activity, but the patient is still engaged in sports at the age of 16 years. Cramps, myalgia and fatigue were not common among the other patients. The symptoms typically started with tripping caused by foot dorsiflexor weakness, which over years progressed to foot drop. In addition, difficulties when walking uphill or standing up were also noted at disease onset. While the majority of patients had disease onset in their 20s, it was noted that all three patients homozygous for the common variant (nos 02, 05 and 13) had an onset of symptoms in their 40s (Table 3). On the other hand, the three patients compound heterozygous for both founder variants (nos 01, 08 and 14) were among those with the earliest onset of symptoms (Table 3).
The facial and bulbar muscles were not affected. Scapular winging and weakness of the shoulder girdle muscles were present in about half of the patients, and were usually mild. Other muscles of the upper limbs were rarely affected, with the exception of mild weakness of the elbow extensors that was noted in four patients (Table 2). Predominant lower limb involvement was a distinctive characteristic of the disease in our patient cohort (Figure 3). A common feature was prominent muscle wasting of the anterior compartment of the lower legs. This was rarely the only region involved (4/14), more often it was combined with wasting of the posterior compartment of the lower legs and of the hamstring muscles (9/14). Two of those patients showed general wasting of the lower limbs (Table 2). Muscle hypertrophy was not detected. Muscle strength was most severely diminished in the distal muscles of the lower limbs, with foot and toe dorsiflexors being most severely affected. Plantar flexors were only mildly affected in the majority of patients, while more than half of the patients had proximal weakness of their lower limbs, mainly affecting the hip muscles (8/14) and the knee flexors (7/14) and extensors (5/14; Table 2). Contractures of the AT were present in all patients (Table 2). Contractures or hypermobility in other joints were not noted. Asymmetric muscle involvement was present in 5/14 patients. All patients but one, who started using a wheelchair at the age of 46 years (no. 04), remained ambulant, with the oldest patient being 53 years of age (Table 2). There was no notable difference in the pattern or severity of muscle wasting and weakness between patients homozygous for the common variant and compound heterozygous patients. In addition, the only patient carrying the common variant in combination with a missense variant (no.10) presented a similar clinical presentation to the other null compound heterozygous cases, suggesting that this variant is likely pathogenic.

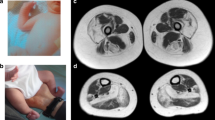
Lower limb involvement in patients with TTN variants. Muscle atrophy is most pronounced in the anterior compartment of the lower legs, but can also be seen in the posterior compartment of the lower legs and in the hamstring muscles. The two upper images show the legs of patient no. 01 and the lower images the legs of patient no. 11.
Normal serum CK levels were noted in six patients, and mildly elevated levels in eight patients (normal value <150 IU/l). EMG showed a myopathic pattern and nerve conduction velocities (NCVs) were normal in all 14 patients. Regular cardiologic examinations were normal in all but one patient (no. 07), who had a mild dilated cardiomyopathy diagnosed at the age of 46 years. Echocardiography showed borderline diastolic and normal systolic dimension of the left ventricle and global hypocontractility of the left ventricle with ejection fraction of 45%. The patient has been treated with bisoprolol 2.5 mg each day and furosemide 40 mg every third day. The last cardiac ultrasound was performed at the age of 53 years – progression was not observed. The respiratory function as measured by spirometry was normal in all patients.
Two patients (nos 06 and 07) reported family members with muscle symptoms. Sister of patient no. 06 has walking difficulties without cardiac symptoms, but she declined clinical examination and genetic analysis. Brother of patient no. 07 had leg weakness and died from cardiac disease at the age of 34 years, but we were not able to obtain more detailed data. There were no muscle and cardiac diseases in family members of other patients, and we did not observe any abnormalities on neurological examination in all those who accepted to be examined.
Muscle biopsy
Seven patients underwent a muscle biopsy. Most biopsies showed end-stage findings with atrophic fibres, lots of fibrous tissue, fatty replacement and an increase in internal nuclei. The presence of vacuoles was also noted in three patients. There were no neurogenic findings. The muscle biopsy of the youngest patient (no. 14) with a short course of the disease showed milder changes with muscle fibre size variation (hypotrophy and occasional atrophy), increase in connective tissue and visible macrophages. Findings in all patients were compatible with muscular dystrophy (Figure 4). No specific immunohistochemical changes were detected and no signs of protein accumulation.

Histological findings in muscles biopsies of patients with TTN variants. Haematoxylin and eosin staining of muscle tissue from patient nos 01, 10 and 14. Biopsies showed an increase in internal nuclei (a), dystrophic changes with significant fibrous and fat tissue replacement, but without predominance of muscle fibre types (b). For comparison, the biopsy of the youngest patient shows minor fibre size variation and an increase in connective tissue only (c).
Muscle MRI
Muscle MRI of the thighs and lower legs was performed in six patients (Figure 5). The youngest patient (no. 14) had a completely normal muscle MRI in all applied sequences. The other five patients showed severely affected anterior compartments of the lower legs (5/5), with fatty replacement of muscle tissue. In all but one patient this region was more severely affected than the posterior compartment. The most severely affected muscle in the anterior compartment was the tibialis anterior (5/5) followed by the extensor digitorum longus muscle (3/5). The peroneal muscles were involved in three patients. Fatty replacement in the posterior compartment was far less prominent and was only seen in three of the six patients, affecting the gastrocnemius muscles, the soleus and also the tibialis posterior muscle (1/3). One patient (no. 01) showed very notable asymmetry of calf muscle involvement, with the tibialis anterior, peroneus longus and gastrocnemius muscles being affected on one side, while only the tibialis posterior was unaffected on the other. The hamstring muscles were the most frequently involved muscles of the thighs. Fatty replacement was most commonly seen in the biceps femoris (4/5), followed by semimembranosus (3/5), and semitendinosus muscle (3/5). Several patients also showed involvement of the femoral quadriceps muscles, while a lesser degree of degeneration was observed in the adductor longus of two patients and in the sartorius muscle of another patient. In conclusion, both proximal and distal muscle involvement was seen on T1w axial MR images. The hamstring muscles were the most severely affected muscles of the thighs, while the anterior compartment of the lower legs showed more severe pathology than the posterior compartment.

Thigh and calf muscles of six patients with titinopathy assessed by T1w MRI. For all patients one axial image of the lower legs and two images of the thigh muscles are shown. (a) In patient no. 01, the hamstring muscles were severely affected (asterisk) and there was notable asymmetry in pathology of the calf muscles (arrow). (b) Patient no. 03. showed both hamstring (asterisk) and femoral quadriceps muscle involvement (arrow). Both anterior and posterior compartments of the lower legs were asymmetrically affected. (c) There was marked atrophy in an obese patient. (d) In patient no. 09, the semimembranosus muscle was mildly affected (arrow), while in the lower legs the tibialis anterior, extensor digitorum longus and peroneus muscles were severely affected (asterisk) and the posterior compartment spared. (e) Patient no.13 showed a typical pattern of affected hamstring (asterisk) and tibialis anterior muscles (arrow). (f) The youngest patient, no. 14, with short disease course and normal MRI of muscles.
Discussion
We report on a cohort of patients with distal myopathy of Serbian ancestry. Our results show that recessive variants in the TTN gene, notably the common founder variant c.107635C>T, p.(Gln35879Ter), are responsible for the fairly uniform and recognisable phenotype resembling that of TMD.6
Our findings support an autosomal recessive mode of inheritance for the distal myopathy associated with the common TTN variant c.107635C>T. Segregation analysis within families of our patients showed that a heterozygous state of the c.107635C>T variant is insufficient to cause the disease. We showed that other novel TTN variants found in a compound heterozygous state are pathogenic in combination with the common variant. The common variant c.107635C>T is located in the Mex5 (362) exon, encoding a part of the M-line region of titin, while other observed variants are located in the Mex1 (358) exon, encoding also a part of the M-line region, and in exons encoding either A- or I-band regions of titin (Table 1). To our knowledge, only one Mex5 variant associated with TMD has been reported in two generations (mother and son) of French family C.9 Originally it was described as a dominant variant probably arising de novo in the mother,9 but later two different second TTN variants, located in exons encoding the A-band region of titin, were identified in these patients.12 Only a few variants outside the Mex5 and Mex6 exons have been associated with TMD or LGMD2J, and all have been described as a second variant in patients bearing Mex5 or Mex6 variants and presenting with more severe, complex or unusual phenotypes,12 although many of them have been reported in other titinopathies.3 It has been suggested that sequencing of the last six TTN exons (Mex1–6), encoding the M-line part of titin, should be performed in patients with a TMD phenotype,1 which is in accordance with the identification of the common variant in Mex5 and the three variants in Mex1 in our cohort.
The observed common haplotype shared by the chromosomes harbouring the c.107635C>T variant, with the 1 Mb core haplotype, favours the hypothesis of a single founder event for this variant. The estimated age of c.107635C>T variant in our sample is about 800–1000 years. Although speculative, this estimation suggests that the most recent common ancestor probably lived in Medieval Serbia. As all analysed patients are of Serbian origin from present-day countries of Serbia and Bosnia and Herzegovina, it can be speculated that the founder variant might have arisen around the time when the majority of the Medieval Serbian principalities were united in a state called Raška (Rascia), which then resulted in the establishment of Serbia in the Balkans. Considering a broad 95% CI and inability to estimate the true variant age by the applied single locus method, it may not be excluded that the founder event might have happened earlier, but probably after the South Slavs (including Serbs) settled in the Balkan during the Early Middle Age. The chromosomes with the second founder variant c.103360delG share the more extended common haplotype (at least 7.4 Mb) in comparison with the common c.107635C>T variant, suggesting that it is a more recent founder event. It can be expected for both founder variants, particularly for the common one, to be found elsewhere in the Balkan countries and possibly also in other populations.
The apparent high frequency of the founder c.107635C>T variant, associated with a rare disease resembling TMD in a country with a small population size, probably makes it the most common cause of distal myopathy in Serbian patients. This finding, together with the better described phenotype, may facilitate genetic testing, which should be incorporated at an early stage in diagnostic algorithms for Serbian patients with distal myopathy.
Salient phenotypical features in our cohort are predominant lower limb involvement and prominent weakness of distal muscles, especially the ankle and toe dorsiflexors. Despite the predominance of distal involvement, mild to moderate weakness of proximal leg muscles was observed in most patients and should be added as one of the hallmarks of the disease. Asymmetry was not an uncommon feature. Sparing of hand muscles despite the disease duration is worth mentioning as well. Involvement of facial or bulbar muscles was not observed. Our results did not suggest gender predominance or differences in disease course or severity between female and male patients.
Clear similarities with TMD suggest that the disease in our patients represents one part of the spectrum of the same disorder. On the other hand, some differences should be highlighted. TMD is an autosomal dominant disorder,6 and homozygosity for the variant shows a different, more severe phenotype of LGMD2J.7 Our homozygous patients for the common variant c.107635C>T did not show specific phenotypical features or differences in disease severity. They did show later onset of the disease, in their 40s, which is similar to the onset described for TMD.
For the majority of our patients disease onset was in their 20s. In addition, affection of the hip and shoulder girdle, not typically seen in TMD, was one of the prominent phenotypical features in our cohort. Previously atypical TMD phenotypes were explained by compound heterozygosity in patients with the FINmaj TTN variant.12 There is a lot more to be understood on how and why different TTN variants contribute to the phenotype. The molecular mechanisms underlying distal myopathies caused by TTN variants are far from fully understood, which makes interventional therapy strategies more challenging.
All but one of our patients remained ambulant throughout the follow-up period. This form of titinopathy is unlikely to result in wheelchair dependency before the sixth decade in the majority of patients, but bearing in mind that the oldest patient was 53 years old when last seen, further follow-up will be needed. Life expectancy seems not to be diminished, considering that no heart or respiratory muscle involvement has been observed, except for cardiomyopathy in one patient.
In addition to the clinical diagnosis, a normal to mildly elevated serum CK level, a myopathic EMG, normal NCVs, normal cardiac and respiratory assessments and a dystrophic muscle biopsy are all helpful, although unspecific tools, in establishing the diagnosis.
Muscle MRI could be a helpful diagnostic tool. The selective pattern of involvement can vary between patients, but we found prominent muscle pathology in the anterior compartment of the lower legs together with involvement of the calf and of the hamstring muscles. This pattern is similar to the MRI pattern described in patients with autosomal dominant TMD.1 However, we noted that the extent and localisation of muscle pathology was variable depending on the stage of the disease and that fatty replacement of muscle tissue was also seen in some muscles not typical for TMD, such as the peroneal muscles.
In this study, we presented a distinct distal myopathy phenotype found in a Serbian patient cohort, which may facilitate the diagnosis of this condition in other patients. Population studies are still to be performed, but it is already clear that the common TTN founder variant explains a sizable portion of distal myopathy patients from Serbia and may represent the most common single cause of distal myopathy in patients of Serbian origin. It is now important to establish prevalence data of this disease in Serbia, as well as in other surrounding Balkan countries to adjust the diagnostic algorithm. One of the earliest diagnostic steps in patients with distal myopathy should be variant screening of the common founder TTN variant.
References
Udd B Dystal myopathies; in Karpati G, Hiltin-Johnes D, Bushby K, Griggs C (eds): Disorders of Voluntary Muscle. Cambridge University Press: Cambridge, UK, 2010, pp 35–40.
Udd B : Molecular biology of distal mucular distrophies – sarcomeric proteins on top. Biochim Biophys Acta 2007; 1772: 145–158.
Chauveau C, Rowell J, Ferreiro A : A rising titan: TTN review and mutation update. Hum Mutat 2014; 35: 1046–1059.
Lek M, MacArthur DG : The challenge of next generation sequencing in the context of neuromuscular diseases. J Neuromuscul Dis 2014; 1: 135–149.
Biancalana V, Laporte J : Diagnostic use of massively parallel sequencing in neuromuscular diseases: towards an integrated diagnosis. J Neuromuscul Dis 2015; 2: 193–203.
Udd B, Partanen J, Halonen P et al: Tibial muscular dystrophy. Late adult-onset distal myopathy in 66 Finnish patients. Arch Neurol 1993; 50: 604–608.
Hackman P, Vihola A, Haravuori H et al: Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet 2002; 71: 492–500.
van den Bergh PY, Bouquiaux O, Verellen C et al: Tibial muscular dystrophy in a Belgian family. Ann Neurol 2013; 54: 248–251.
Hackman P, Marchand S, Sarparanta J et al: Truncating mutations in C-terminal titin may cause more severe tibial muscular dystrophy (TMD). Neuromuscul Disord 2008; 18: 922–928.
Udd B : Limb-girdle type muscular dystrophy in a large family with distal myopathy: homozygous manifestation of a dominant gene? J Med Genet 1992; 29: 383–389.
Pollazzon M, Suominen T, Penttilä S et al: The first Italian family with tibial muscular dystrophy caused by a novel titin mutation. J Neurol 2010; 257: 575–579.
Evilä A, Vihola A, Sarparanta J et al: Atypical phenotypes in titinopathies explained by second titin mutations. Ann Neurol 2014; 75: 230–240.
Edström L, Thornell LE, Albo J, Landin S, Samuelsson M : Myopathy with respiratory failure and typical myofibrillar lesions. J Neurol Sci 1990; 96: 211–228.
Girdea M, Dumitriu S, Fiume M et al: PhenoTips: Patient Phenotyping Software for clinical and research use. Hum Mutat 2013; 34: 1057–1065.
Köhler S, Doelken SC, Mungall CJ et al: The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucl Acids Res 2014; 42: D966–D974.
Personius KE, Pandya S, King WM, Tawil R, McDermott MP The FSH DY Group:: Facioscapulohumeral dystrophy natural history study: standardization of testing procedures and reliability of measurements. Phys Ther 1994; 74: 253–263.
Hollingsworth KG, de Sousa PL, Straub V, Carlier PG : Towards harmonization of protocols for MRI outcome measures in skeletal muscle studies: consensus recommendations from two TREAT-NMD NMR workshops, 2 May 2010, Stockholm, Sweden, 1–2 October 2009, Paris, France. Neuromuscul Disord 2012; 22 (Suppl 2): S54–S67.
Willis TA, Hollingsworth KG, Coombs A, Sveen ML, Andersen S, Stojkovic T : Quantitative magnetic resonance imaging in limb-girdle muscular dystrophy 2I: a multinational cross-sectional study. PLoS One 2014; 9: e90377.
Mercuri E, Pichiecchio A, Counsell S et al: A short protocol for muscle MRI in children with muscular dystrophies. Eur J Paediatr Neurol 2002; 6: 305–307.
Serre JL, Simon-Bouy B, Mornet E et al: Studies of RFLP closely linked to the cystic fibrosis locus throughout Europe lead to new considerations in populations genetics. Hum Genet 1990; 84: 449–454.
Slatkin M, Rannala B : Estimating allele age. Annu Rev Genomics Hum Genet 2000; 1: 225–249.
Acknowledgements
We thank the patients and their relatives for their willingness to cooperate in this study. We would also like to thank Mojgan Reza and Dan Cox for technical support, and Dr Monica Ensini for managing the MYO-SEQ study. MYO-SEQ is funded by Sanofi Genzyme, Ultragenyx Pharmaceutical, the LGMD2I Research Fund, the Kurt+Peter Foundation, the LGMD2D Foundation and the Samantha J Brazzo Foundation. The study was also supported by the Medical Research Council UK (reference G1002274, Grant No. ID 98482), by the European Union Seventh Framework Programme (FP7/2007–2013) under Grant Agreement Nos 305444 (RD-Connect) and 305121 (Neuromics) to VS and HL and by the Ministry of Education, Science and Technological Development, Republic of Serbia (Grants Nos 175083 and 173016). Dr Jelena Nikodinovic Glumac has received 6 months Fellowship from the European Academy of Neurology, which made her work on this study possible.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Perić, S., Glumac, J., Töpf, A. et al. A novel recessive TTN founder variant is a common cause of distal myopathy in the Serbian population. Eur J Hum Genet 25, 572–581 (2017). https://doi.org/10.1038/ejhg.2017.16
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.16
This article is cited by
-
Exploring TTN variants as genetic insights into cardiomyopathy pathogenesis and potential emerging clues to molecular mechanisms in cardiomyopathies
Scientific Reports (2024)
-
Aberrant mRNA processing caused by splicing mutations in TTN-related neuromuscular disorders
Journal of Human Genetics (2023)
-
Whole-body muscle MRI of patients with MATR3-associated distal myopathy reveals a distinct pattern of muscular involvement and highlights the value of whole-body examination
Journal of Neurology (2020)
-
Detection of variants in dystroglycanopathy-associated genes through the application of targeted whole-exome sequencing analysis to a large cohort of patients with unexplained limb-girdle muscle weakness
Skeletal Muscle (2018)
