
Abstract
There are at least 38 mutant genes known to be associated with the ichthyosis phenotypes, and autosomal recessive congenital ichthyosis (ARCI) is a specific subgroup caused by mutations in 13 different genes. Mutations in some of these genes, such as CERS3 with only two previous reports, are rare. In this study, we identified mutations in candidate genes in consanguineous families with ARCI with a next generation sequencing (NGS) array that incorporates 38 ichthyosis associated genes. We applied this sequencing array to DNA from 140 ichthyosis families with high prevalence of consanguinity. Among these patients we identified six distinct, previously unreported mutations in CERS3 in six Iranian families. These mutations in each family co-segregated with the ichthyosis phenotype. The patients demonstrated collodion membrane at birth, acrogeria, generalized scaling, and hyperlinearity of the palms and soles. The presence of a significant percentage of CERS3 mutations in our cohort depicts a marked difference between the etiology of ichthyoses in genetically poorly characterized regions and well-characterized western populations. Also, it shows that rare alleles are more prevalent in the gene pool of consanguineous populations and emphasizes the importance of these population studies for better understanding of ichthyosis pathogenesis.
Similar content being viewed by others

Introduction
Generalized Mendelian disorders of cornification are caused by mutations in genes that play a role in epidermal differentiation and barrier function, and there are as many as 38 distinct genes known to be associated with the ichthyosis phenotypes.1 This group of disorders is highly heterogeneous both in phenotypic presentation, characterized by scaling and hyperkeratosis, as well as in genetics, manifesting with autosomal dominant, autosomal recessive or X-linked recessive inheritance. Autosomal recessive congenital ichthyosis (ARCI) is a specific subgroup of inherited ichthyoses, and mutations in as many as 13 distinct genes important for keratinization and lipid metabolism have been described in ARCI; these genes include ABCA12, ALOXE3, ALOX12B, CASP14,CERS3, CYP4F22, LIPN, NIPAL4, PNPLA1, SDR9C7,ST14,SULT2B1, and TGM1.2 Mutations in some of these genes, for example TGM1, are relatively common while mutations in other genes, including PNPLA1, LIPN, and CERS3, have been reported only in a limited number of families. For example, only two reports on mutations in CERS3 have been published on consanguineous Turkish and Tunisian families.3, 4 Here we ascertained a large cohort of 140 distinct Iranian families affected by non-syndromic and syndromic forms of ichthyosis. To disclose the molecular pathology of ichthyoses in Iran, a country of ~80 million inhabitants with high prevalence of customary consanguineous marriages, we designed a disease-targeted next generation sequencing (NGS) panel covering 38 genes associated with different forms of ichthyoses. In this study, we found six different, previously unreported, homozygous mutations in CERS3 in six consanguineous Iranian families.
Materials and methods
Patients and clinical phenotyping
This study was approved by the Institutional Review Board of the Pasteur Institute of Iran, and all subjects and parents or guardians of under-aged patients gave written informed consent to participate in this research. In this study, 140 extended families affected by non-syndromic and syndromic forms of ichthyosis, diagnosed in various medical centers in Iran were subjected to genetic analysis. In 112 of these families, specific mutations were disclosed in 16 distinct genes. In this report, we focus on clinical and genetic characteristics of patients representing six families with mutations in CERS3.
Next generation sequencing and data analysis
DNA was extracted from peripheral blood samples by a kit (QIAamp Blood Maxi Kit; Qiagen, Valencia, CA, USA) or by salting out method.5 DNA concentration was measured using a Qubit 2.0 fluorometer (Life Technologies, Carlsbad, CA, USA). Target enrichment was performed using the TruSeq Custom Amplicon kit (Illumina Inc., San Diego, CA, USA). DesignStudio (Illumina Inc.) was used for library design. All coding exons, at least 20 bp of the intron at each intron–exon boundary, and up to 50 bp of 3′-UTRs were targeted. The targeted NGS sequencing panel contained 38 genes (ABCA12, ABHD5, AGPS, ALDH3A2, ALOX12B, ALOXE3, AP1S1, ARSE, CERS3, CLDN1, CYP4F22, EBP, ELOVL4, GJB2, GJB3, GJB4, GJB6, KRT1, KRT10, KRT2, KRT9, LIPN, LOR, NIPAL4, PEX7, PHYH, PNPLA1, PNPLA2, POMP, SLC27A4, SNAP29, SPINK5, ST14, STS, TGM1, TGM5, VPS33B, and ZMPSTE24) divided into 351 targets covered by 558 amplicon probes which were designed to cover 99% of targeted bases. A total of 93.2% of the reads were aligned to the human genome, with the mean coverage of the target region being 493X. In addition, only 0.4% of bases of the target region were not covered by any sequence read, indicating that 99.6% of all target region bases were sequenced at least once. Genomic DNA from 140 probands each representative of a distinct extended family and 2 Illumina controls were multiplexed using dual indexing with 12 primary indexes and eight secondary indexes. Dual indexed samples were normalized to be equimolar and pooled together following manufacturer’s recommendations. The pool was sequenced on a single MiSeq flow-cell (Illumina Inc.). Reads were paired-end at 2 × 225 nt with dual indexes, and a total of 8.49 Gbp were generated. Variants were called with GATK HaplotypeCaller.
Sanger sequencing
PCR was performed using Taq polymerase (Qiagen) according to the manufacturer's instructions. Amplification of the CERS3 gene was performed with four pairs of newly designed primers (sequences available upon request), spanning all four exons harboring mutations found by NGS and their flanking intronic sequences. The PCR products were bidirectionally sequenced using 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA). The mutation positions are reported in reference to NG_042826.1 (genomic), NM_178842.3 (cDNA) and NP_849164.2 (protein).
Results
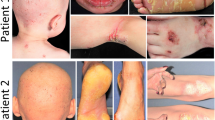
In this study, we identified six previously unreported mutations in the CERS3 in six families with ARCI. The affected family members demonstrated collodion membrane at birth, generalized scaling consisting of fine or large scales, acrogeria, hyperlinearity of the palms and soles, and ectropion later in life (Figure 1). Histopathology was consistent with ichthyosis demonstrating compact hyperkeratosis, focal hypergranulosis, and regularly acanthotic epidermis (Figure 1k). For detailed clinical description of the phenotype in these families, see Supplementary Material online. Three families, nos. 2, 5, and 6, harbored homozygous missense variants, c.685C>T, (p.(Arg229Cys)); c.915C>A, (p.(Asp305Lys)), and c.686C>T, (p.(Arg229His)), respectively (Figures 2b, e, and f). (For sequence variants and the corresponding wild-type sequences, see Case Reports in Supplementary Material online). Bioinformatics predictions of these variants with mutation taster, PolyPhen2, FIS, GVGD, PROVEAN, and SIFT programs, predicted them to be pathogenic (Supplementary Table S1 online). In two independent families (nos. 3 and 4) of different ethnicities and from different regions of Iran, a homozygous nonsense variant, c.30G>A, (p.(Trp10Ter)), was identified. One of the families (no. 1) harbored two different homozygous sequence variants in CERS3. One variant consisted of substitution of three nucleotides, AAA, encoding Lys131 in the control population, by GCC leading to p.Lys131Ala substitution which was suggested to be pathogenic by prediction programs. In addition, these patients harbored a homozygous in-frame c.401_403del, p.(Glu134del) deletion mutation (Figure 2a). These mutations in each family co-segregated with the ichthyosis phenotype, the parents being heterozygous carriers. In silico analysis of the missense variant p.(Lys131Ala) predicted it as damaging, but with limited existing data about this family with two variants, we were not able to determine whether the phenotype resulted from either one of these mutations alone or from a combination of both of them. All variants were confirmed by bidirectional Sanger sequencing and submitted to LOVD: www.lovd.nl/cers3 (patient IDs: 104974-978 and 105000-01; variant IDs: 170861-64 and 170891-2). None of these genomic variants have been reported previously, and they were absent in 119,654 alleles in control population (ExAC.broadinstitute.org) and in the Greater Middle East (GME) Variome Project (igm.ucsd.edu/gme).

Clinical features and histopathology of patients with CERS3 mutations. Note generalized fine scaling and hyperlinearity of the palms and soles. The patients also showed acrogeria-like features on the backs of the hands (a–j). Characteristic histopathology findings of skin biopsy demonstrated compact hyperkeratosis, focal hypergranulosis, and regularly acanthotic epidermis, with normal dermis without inflammatory cell infiltrate (k). (H&E stain, original magnification × 20).

Pedigrees and mutations in CERS3 in six families with ARCI. The probands are identified by arrowheads; the mutations are identified by red arrows. For the corresponding wild-type sequences, see Supplementary Material online. Note the consanguinity in each family (a–f). The positions of the mutations identified in CERS3 at protein level (g) and within the gene (h). CERS has six transmembrane α-helical domains (yellow) traversing the nuclear membrane with the start and end amino acid positions indicated. The gene consists of 13 exons, the translation initiation codon ATG being in exon 4. The sizes of the exons are shown in parentheses; the introns are not drawn into scale. The sequences corresponding to Homeobox Domain (DNA binding) and thin-layer chromatography (TLC) domain are indicated.
Discussion
In this study, we report six previously unreported variants in CERS3 in six extended families among the 140 families tested (4.3%) affected by non-syndromic, autosomal recessive congenital ichthyosis (OMIM#615023). Combining a large cohort of patients with ichthyosis, born to consanguineous parents, with the utilization of NGS panel targeting 38 ichthyosis associated genes allowed us to expand genotypic spectrum of CERS3.3, 4, 6 Our study now increases the total number of reported CERS3 mutations in ARCI from 2 to 8, scattered along the entire gene (Figures 2g and h). Our study also reports a stop codon mutation which resided in exon 4, just 10 amino acids upstream from the Met1. Interestingly, examination of the clinical phenotype in patients with CERS3 mutations did not reveal notable differences in the severity between the patients with this variant, p.(Trp10Ter), and those with missense substitutions along the polypeptide. Overall, the clinical phenotype in the nine Iranian patients examined in this study was relatively mild (Figure 1), however, in three of the families there was a history of miscarriages, and in two families (nos. 2 and 5), two children died at an early age due to complications of ichthyosis.
Consanguineous marriage is customary in different geographic regions, specifically in the Middle Eastern and North and Sub-Saharan African countries.7 It is estimated that at least 8.5% of children have consanguineous parents.8 So far, all disclosed CERS3 mutations were homozygous and belong to the families from countries with high rate of customary consanguineous marriages, including Iran, Turkey, and Tunisia with consanguinity rates of 38.5, 21.0, and 21.5%, respectively.9, 10, 11 In addition to the degree of relationship between the parents, the increase in risk of having an affected child with autosomal recessive disease born to consanguineous parents depends on population frequency of the disease allele in inversely proportional order. The less common the disease allele causing the disorder is in the gene pool, the greater the probability that the parents of an affected individual are consanguineous.
CERS3 mutations are examples of extremely rare autosomal recessive human ‘knockout’ alleles with predicted loss-of-function (pLoF).12 So far, all reported patients including our cases in this study had autosomal recessive genotypes and consanguineous parents. We previously reported similar results in the same Iranian population for PNPLA1, FERMT1, CMG2, and KRT14 as causal genes for ARCI type 10 (OMIM # 615024), Kindler syndrome (OMIM # 173650), hyaline fibromatosis syndrome (OMIM # 228600), and autosomal recessive epidermolysis bullosa simplex (OMIM # 601001), respectively. In these extremely rare Mendelian disorders, all reported variants were homozygous, and all subjects had consanguineous parents.13, 14, 15, 16
In contrast to very rare Mendelian disorders as mentioned above, the rate of parental consanguinity is lower in more prevalent autosomal recessive single gene diseases. For instance, 92% of the patients with recessive dystrophic epidermolysis bullosa (RDEB) (OMIM # 226600) had consanguineous parents.17 Similarly, in an Iranian cohort of 515 patients affected by a heterogeneous group of primary immunodeficiency disorders, the average consanguineous marriage rate among parents of ataxia-telangiectasia patients was 81%.18 Also, the consanguinity rate among the parents of autosomal recessive non-syndromic deafness patients from Iran and Turkey was 73 and 70%, respectively.19 Therefore, the rarer a particular disease is in a population, the more likely the parents are to be consanguineous as initially was surmised by Archibald Garrod in the case of alkaptonuria more than one century ago and corroborated by recent studies.
In summary, application of a NGS array targeting 38 ichthyosis associated genes in a large cohort of ichthyosis patients from a consanguineous population allowed us to identify previously unreported mutations in CERS3 which brings the total number of mutations reported in this gene to eight.
References
Baden HP, Digiovanna JJ Ichthyosiform dermatoses. In:Rimoin DL(eds:)Emery and Rimoin's Principle and Practice of Medical Genetics. Academic Press: Philadelphia, 2013,pp 1–26.
Richard G, Bale SJ Autosomal recessive congenital ichthyosis; in: Pagon R, Adam MP, Ardinger HH et al(eds)GeneReviews. University of Washington: Seattle (WA), 2001, January 10, 2001; Last Update: August 28, 2014 edn.
Radner FP, Marrakchi S, Kirchmeier P et al: Mutations in CERS3 cause autosomal recessive congenital ichthyosis in humans. PLoS Genet 2013; 9: e1003536.
Eckl KM, Tidhar R, Thiele H et al: Impaired epidermal ceramide synthesis causes autosomal recessive congenital ichthyosis and reveals the importance of ceramide acyl chain length. J Invest Dermatol 2013; 133: 2202–2211.
Miller SA, Dykes DD, Polesky HF : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Jennemann R, Rabionet M, Gorgas K et al: Loss of ceramide synthase 3 causes lethal skin barrier disruption. Hum Mol Genet 2012; 21: 586–608.
Hamamy H, Antonarakis SE, Cavalli-Sforza LL et al: Consanguineous marriages, pearls and perils: Geneva International Consanguinity Workshop Report. Genet Med 2011; 13: 841–847.
Modell B, Darr A : Science and society: genetic counselling and customary consanguineous marriage. Nat Rev Genet 2002; 3: 225–229.
Saadat M, Ansari-Lari M, Farhud DD : Consanguineous marriage in Iran. Ann Hum Biol 2004; 31: 263–269.
Ben Halim N, Ben Alaya Bouafif N, Romdhane L et al: Consanguinity, endogamy, and genetic disorders in Tunisia. J Community Genet 2013; 4: 273–284.
Tuncbilek E, Koc I : Consanguineous marriage in Turkey and its impact on fertility and mortality. Ann Hum Genet 1994; 58: 321–329.
Saleheen D, Natarajan P, Armean IM et al: Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature 2017; 544: 235–239.
Vahidnezhad H, Youssefian L, Saeidian AH et al: Gene-targeted next generation sequencing identifies PNPLA1 mutations in patients with a phenotypic spectrum of autosomal recessive congenital ichthyosis: the impact of consanguinity. J Invest Dermatol 2017; 137: 678–685.
Youssefian L, Vahidnezhad H, Barzegar M et al: The Kindler syndrome: a spectrum of FERMT1 mutations in Iranian families. J Invest Dermatol 2015; 135: 1447–1450.
Youssefian L, Vahidnezhad H, Aghighi Y et al: Hyaline fibromatosis syndrome: a novel mutation and recurrent founder mutation in the CMG2/ANTXR2 gene. Acta Derm Venereol 2017; 96: 108–109.
Vahidnezhad H, Youssefian L, Saeidian AH et al: KRT5 and KRT14 mutations in epidermolysis bullosa simplex with phenotypic heterogeneity, and evidence of semi-dominant inheritance in a multiplex family. J Invest Dermatol 2016; 136: 1897–1901.
Vahidnezhad H, Youssefian L, Zeinali S et al: Dystrophic epidermolysis bullosa: COL7A1 mutation landscape in a multi-ethnic cohort of 152 extended families with high degree of customary consanguineous marriages. J Invest Dermatol 2017; 137: 660–669.
Rezaei N, Pourpak Z, Aghamohammadi A et al: Consanguinity in primary immunodeficiency disorders; the report from Iranian Primary Immunodeficiency Registry. Am J Reprod Immunol 2006; 56: 145–151.
Bademci G, Foster J 2nd, Mahdieh N et al: Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. Genet Med 2016; 18: 364–371.
Acknowledgements
We would like to thank Kaveh Khademhossein and Maryam Abiri for assistance in sample collection. Carol Kelly assisted in manuscript preparation. This study is in partial fulfillment of the PhD Thesis of Leila Youssefian.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Youssefian, L., Vahidnezhad, H., Saeidian, A. et al. Autosomal recessive congenital ichthyosis: CERS3 mutations identified by a next generation sequencing panel targeting ichthyosis genes. Eur J Hum Genet 25, 1282–1285 (2017). https://doi.org/10.1038/ejhg.2017.137
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.137
This article is cited by
-
New developments in the molecular treatment of ichthyosis: review of the literature
Orphanet Journal of Rare Diseases (2022)
-
Mutation in ALOX12B likely cause of POI and also ichthyosis in a large Iranian pedigree
Molecular Genetics and Genomics (2020)
-
Exacerbation of ichthyosis vulgaris phenotype by co-inheritance of STS and FLG mutations in a Chinese family with ichthyosis: a case report
BMC Medical Genetics (2018)
-
Molecular genetics of a cohort of 635 cases of phenylketonuria in a consanguineous population
Journal of Inherited Metabolic Disease (2018)
