
Abstract
About one third of patients with rhabdoid tumors (RT) harbor a heterozygous germline variant in SMARCB1. Molecular diagnosis therefore keeps a crucial place in the diagnosis of RT, and genetic counseling should be systematically recommended. However, immunohistochemistry has progressively replaced molecular tools to assess the status of SMARCB1 in tumors; the necessity of analyzing SMARCB1 status in the tumor may thus be less considered by neuropathologists and pediatric neuro-oncologists. In the present manuscript as aforementioned, we report on two patients with bifocal RT in the first month of life and in whom no germline variant was initially found in the SMARCB1 coding sequence. Careful analysis of SMARCB1 status in the tumors revealed that only one of the two inactivating hits was found in the coding sequence. By sequencing the tumor cells RNA, we were able to detect an insertion with an abnormal sequence, due to the same intronic variant of SMARCB1, which led to the exonisation of the first intron. This cryptic variant was absent in the germline DNA of both patients. Of note, we previously reported one patient with the same deep intronic variant in the germline in a soft tissue RT. To our mind, this additional report on two patients clearly demonstrates that this intronic variant is a new hotspot that should now be systematically added to the germline screening of SMARCB1. We therefore recommend searching for and cautiously interpreting germline analysis if SMARCB1 has not been extensively studied in the tumor.
Similar content being viewed by others


Introduction
Atypical Teratoid and Rhabdoid Tumors (AT/RTs) represent the central nervous system form of rhabdoid tumors (RT). They are clinically aggressive malignancies most frequently diagnosed in young children with a median age of 2 years. In spite of their pleomorphic features, they all share the same genetic driver event, the biallelic inactivation of the SMARCB1 tumor suppressor gene which is somatically acquired in tumors. A subset of these tumors occurs in a context of genetic predisposition (Rhabdoid predisposition syndrome, RPS) due to germline variants in the SMARCB1 gene. We report herein on two patients harboring a germline intronic variant in the SMARCB1 gene evidenced by molecular analysis of tumor cells cDNA and revealing a RPS.
Subjects and methods
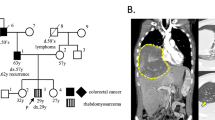
A 4-month-old boy presented with a progressive macrocephaly for which magnetic resonance imaging (MRI) of the skull was performed and revealed a mass located on the cerebellar falx (Figure 1a). Histopathological analysis of biopsy material showed a poorly differentiated monomorphous tumor (Figure 1c) made of round-shaped tumor cells with scant cytoplasm and loss of nuclear immunoreactivity for INI1 antibody (Figure 1d). Therefore, a diagnosis of AT/RT, WHO grade IV, was made.1 An abdominal computed tomography scan evidenced a lesion within the left kidney suggestive of a RT (Figure 1b) in a context of RPS. The patient died 6 months after the initial diagnosis.

Radiological and histopathological findings of patient 1 (a–d) and 2 (e–h) tumors: sagittal FLAIR magnetic resonance imaging showing hydrocephalus caused by a large hyperintense falx cerebellar lesion compressing the aqueduct of Sylvius (a). Computerized tomodensitometry scan displaying a well-circumscribed heterogeneous lesion of the right kidney on T1 sequence after injection of gadolinium (arrow) (b), composed of small and poorly differentiated cells (c; HE, × 380 magnification); loss of nuclear INI1 expression specifically observed in the tumor cells with preserved immunostaining in normal endothelial cells (d; × 380 magnification); axial magnetic resonance imaging showed an enhancing large hyperintense cerebellar lesion (e) and a second lesion within the left foramen of Monro on T1 sequence after injection of gadolinium (f); composed of poorly differentiated cells with typical features of rhabdoid cells in a part of them (g; HE, × 380 magnification); loss of the nuclear INI1 expression in the tumor cells (h; × 380 magnification).
Another boy of 8-month-old of age was referred in emergency owing to vomiting and progressive enlargement of the head. MRI displayed two voluminous contrast enhancing solid and cystic tumor masses, the first being protruding into the fourth ventricle (Figure 1e) and the second located in the left foramen of Monro (Figure 1f). A subtotal resection of the cerebellar tumor was performed. Microscopic examination displayed a poorly differentiated tumor proliferation, containing small rounded cells with large eosinophilic cytoplasms and nuclear loss of INI1 expression on immunohistochemical analysis (Figures 1g and h), that led to the diagnosis of AT/RT, WHO grade IV.1 The patient was started on chemotherapy (vincristine and doxorubicin). One month later, MRI revealed progression of the supratentorial tumor. The patient died 13 months after the initial diagnosis.
Despite no familial history of RT in these two patients, the synchronous bifocal presentation strongly suggested a germline variant in the SMARCB1 gene, whose all coding exons and splice sites regions were sequenced using the Sanger method from peripheral blood lymphocytes. Large size deletions were then sought using Multiple Ligation-dependent Probe Amplification (Salsa MLPA KIT P258-B1 SMARCB1).
Results and Discussion
These two methods failed to reveal any alteration. DNA was analysed from a frozen tumor sample using the same method. From samples which were rich in tumor cells, molecular analysis revealed a hemizygous deletion of the 9 exons of the gene without any further change in all coding sequences. The strong suspicion of RPS (young age and bifocal tumor) led us to search for other alterations by sequencing the SMARCB1 cDNA in the tumor cells. This analysis showed an insertion of 72 nucleotides located between exons 1 and 2 corresponding to a sequence of the first intron of the SMARCB1 gene that resulted in the formation of a premature termination codon in the tumor of both patients (Figure 2). Sequencing of intron 1 also revealed a variant c.93+559A>G in the genomic DNA, producing a new donor splice site and exonisation of a part of intron 1. This variant was found in the germline DNA of both patients and absent DNA extracted from peripheral blood samples of the parents, confirming the de novo occurrence of the variant and the RPS.

Molecular analysis of tumor DNA showing an insertion of 72 nucleotides between the exons 1 and 2 that resulted in a premature termination codon (TGA) (a); intronic mutation c.93+559A>G (b) evidenced by sequencing of intron 1 producing a new donor splice site and exonisation of a part of intron 1, found also in germline DNA (asterisks) but not in parents’ blood DNA samples (c).
In the majority of RT and AT/RT cases, molecular analysis of tumor DNA reveals a biallelic alteration of SMARCB1. However, MLPA and Sanger sequencing of coding exons may sometimes reveal only one alteration. Therefore, the crucial issue facing us consist in the search for the second genetic event to provide a relevant interpretation from the germline DNA sequencing given the high frequency of germline variants (about 30–35% of patients)2, 3 and actively searching for germline variants is now considered as a good practice.
In cases where only one SMARCB1 alteration is found, a high percentage of normal cell contamination in the samples which can mask a genuine deletion must first be suspected. If the histopathological examination confirms a rate of tumor cells superior to 30%, FISH represents a very useful tool reveal actual homozygous deletions. A second mechanism could result from epigenetic silencing by hypermethylation at the promoter site, but promoter hypermethylation has never been reported so far.4 Finally, cryptic intronic variants that might have been missed by analyses focusing only on exons and introns/exons boundaries must also investigated, in so far as we previously reported on one patient suffering from a soft tissue RT in which the same intronic variant of SMARCB1 was evidenced.5 From this previous case and the two reported cases here, this intronic variant emerges as a hotspot variant in RPS that should be now systematically considered.
In conclusion, instead of illustrating one new cryptic intronic variant, this report highlights the necessity to analyze tumor DNA by different approaches. Physicians and geneticists must keep in mind that the absence of germline variant in the coding sequence may have two different meanings, either a truly normal sequence or a normal coding sequence with an abnormal non-coding sequence. In that perspective, the identification of the two events in the tumor DNA is critical for a relevant interpretation and for the identification of RPS.
References
Judkins AR, Eberhart CG, Wesseling P, Hasselblatt M : Atypical teratoid/rhabdoid tumour; In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK WHO Classification of Tumours of the Central Nervous System. revised 4th edition Lyon, France: International Agency for Research on Cancer, 2016; 209–212.
Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B : Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 1999; 59: 74–79.
Bourdeaut F, Lequin D, Brugières L et al: Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res 2011; 17: 31–38.
Zhang F, Tan L, Wainwright LM, Bartolomei MS, Biegel JA : No evidence for hypermethylation of the hSNF5/INI1 promoter in pediatric rhabdoid tumors. Genes Chromosomes Cancer 2002; 34: 398–405.
Sévenet N, Lellouch-Tubiana A, Schofield D et al: Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype-phenotype correlations. Hum Mol Genet 1999; 8: 2359–2368.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Tauziède-Espariat, A., Masliah-Planchon, J., Brugières, L. et al. Deep intronic hotspot variant explaining rhabdoid tumor predisposition syndrome in two patients with atypical teratoid and rhabdoid tumor. Eur J Hum Genet 25, 1170–1172 (2017). https://doi.org/10.1038/ejhg.2017.115
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.115
This article is cited by
-
Current recommendations for clinical surveillance and genetic testing in rhabdoid tumor predisposition: a report from the SIOPE Host Genome Working Group
Familial Cancer (2021)
-
Malignant rhabdoid tumors originating within and outside the central nervous system are clinically and molecularly heterogeneous
Acta Neuropathologica (2018)
