
Abstract
Autosomal dominant polycystic liver disease (ADPLD) is caused by variants in PRKCSH, SEC63, and LRP5, whereas autosomal dominant polycystic kidney disease is caused by variants in PKD1 and PKD2. Liver cyst development in these disorders is explained by somatic loss-of-heterozygosity (LOH) of the wild-type allele in the developing cyst. We hypothesize that we can use this mechanism to identify novel disease genes that reside in LOH regions. In this study, we aim to map abnormal genomic regions using high-density SNP microarrays to find novel PLD genes. We collected 46 cysts from 23 patients with polycystic or sporadic hepatic cysts, and analyzed DNA from those cysts using high-resolution microarray (n=24) or Sanger sequencing (n=22). We here focused on regions of homozygosity on the autosomes (>3.0 Mb) and large CNVs (>1.0 Mb). We found frequent LOH in PRKCSH (22/29) and PKD1/PKD2 (2/3) cysts of patients with known heterozygous germline variants in the respective genes. In the total cohort, 12/23 patients harbored abnormalities outside of familiar areas. In individual ADPLD cases, we identified germline events: a 2q13 complex rearrangement resulting in BUB1 haploinsufficiency, a 47XXX karyotype, chromosome 9q copy-number loss, and LOH on chromosome 3p. The latter region was overlapping with an LOH region identified in two other cysts. Unique germline and somatic abnormalities occur frequently in and outside of known genes underlying cysts. Each liver cyst has a unique genetic makeup. LOH driver gene BUB1 may imply germline causes of genetic instability in PLD.
Similar content being viewed by others


Introduction
Polycystic liver disease (PLD) is part of the phenotype of two inherited disorders; autosomal dominant PLD (ADPLD) and autosomal dominant polycystic kidney disease (ADPKD). In 83–94% of ADPKD patients, polycystic livers are seen.1, 2 Variants in Protein Kinase C Substrate 80 K-H (PRKCSH), SEC63 homolog (Saccharomyces cerevisiae; SEC63), and Low-density lipoprotein Receptor-related Protein 5 (LRP5) cause ADPLD, and are present in ~25% of cases,3, 4 whereas variants in Polycystic Kidney Disease 1 (PKD1) and Polycystic Kidney Disease 2 (PKD2) are responsible for ADPKD in virtually all cases.5 Protein products of genes underlying PLD are located in the endoplasmatic reticulum (ER) or primary cilium.6, 7 Experimental data favor a genetic interaction network between ER-localized protein products of PRKCSH and SEC63, and cilium-localized PKD1 and PKD2.7, 8 The finding of LRP5 variants in PLD suggests that Wnt signaling may be disrupted downstream of this interaction network. Genes that underlie PLD thus function in distinct organelles and pathways, despite a final common cystogenic effect. Furthermore, ciliopathy-associated genes act outside of the PKD1/PKD2 genetic interaction network,7, 9 and may also cause liver cysts. The search for new genes should therefore not be limited to currently known genomic sites. At a tissue level, PLD appears to be a recessive disease. Complete loss of cyst gene expression from diseased epithelium follows loss-of-heterozygosity (LOH),10, 11, 12, 13, 14, 15 which may be related to cyst genetic instability.16, 17 The proportion of somatic variants varies with the gene that is affected in the germline. Recent studies found that second, somatic variants or LOH occurred in 56/71 liver cysts (79%) from patients with PRKCSH variants,11 in 4/5 (80%) PKD2 variant carriers,18 but only 1/14 cysts (7%) from a patient with a SEC63 variant.10 This indicates LOH incidence depends upon the genetic and phenotypic background. We hypothesize that a ‘two-hit model’ is a general principle for the development of hepatic cysts. Therefore, somatic LOH regions in cyst epithelium may harbor novel candidate PLD-causing genes, which harbor heterozygous germline variants in the respective cases. Considering the genetic interaction network in PLD,7, 8 digenic or transheterozygous variants at two genetic loci may also have a role. Transheterozygous PKD1/PKD2 variants have been described in renal cysts,14, 15 whereby a variant in one cyst gene is succeeded by a variant in a second cyst gene. Cysts with heterozygous variants in PRKCSH and SEC63 continue to express the relevant proteins.10, 11 It is reasonable to hypothesize that transheterozygosity may be another mechanism in hepatic cyst formation. This study aims to determine novel genetic loci that are involved in cystogenesis both at germline and somatic level. To this end, we followed an unbiased approach and assessed copy-number variations (CNVs) and LOH regions in PLD cyst epithelium using a genome-wide high-resolution cytogenetic array analysis.
Methods
Patient material
We obtained DNA from liver cyst cholangiocytes of 23 newly included patients who underwent either laparoscopic cyst fenestration or aspiration sclerotherapy from 2011–2014 because of large cysts. All patients except one were females, and had single or multiple liver cysts. All patients had severe symptoms and the mean age was 54 (range 42–83) years. Seventeen patients had ADPLD, three had ADPKD, and three had solitary or sporadic cysts. Use of this tissue for research was reviewed and approved by the regional ethics review board ‘Commissie Mensgebonden Onderzoek regio Arnhem-Nijmegen’.
Cyst work-up
We isolated cholangiocytes by four methods (Supplementary Figure S1; Table 1). First, as described from 23 previously studied laparoscopy-derived liver cysts (six patients) obtained from 2010 to 2012,18 we collected cells from fresh tissue by ethylenediaminetetraacetic acid detachment. Keratin (KRT)-19 staining indicated the purity of each sample. Second, we collected cells from 30 laparoscopy-derived liver cysts of eight patients from 2012 to 2014. These cells expanded into adult liver organoids using conditions suitable for their expansion.19 Under these conditions, only stem cells with a cholangiocyte-like phenotype expressing KRT19 persisted. DNA from one cyst per patient was studied using high-density SNP microarrays (Affymetrix Cytoscan HD, Santa Clara, CA, USA). DNA from the remaining 22 cysts was used to assess somatic loss of the wild-type allele of heterozygous PRKCSH germline variants by Sanger sequencing. Third, symptomatic cyst patients were referred to our hospital for aspiration sclerotherapy. We collected 68 cyst fluid aspirates from 50 patients in 2011 and 2012. We subjected all samples to centrifugation, KRT19 staining, and fluorescent-activated cell sorting (FACS) of cholangiocytes (Supplementary Methods). This yielded eight additional samples for single-nucleotide variant (SNP) microarray studies. Fourth, we grew eight cultures from 30 aspiration sclerotherapy fluids collected from 2012 to 2014, using conditions suitable for the expansion of adult liver stem cells.19 We obtained cyst fluid and epithelium samples, and stored them in the course of treatment following the Dutch Code for the proper secondary use of human tissue. Use of this tissue for research was reviewed and approved by the regional ethics review board ‘Commissie Mensgebonden Onderzoek regio Arnhem-Nijmegen’.
DNA isolation from cyst cholangiocytes
We isolated DNA from the cyst cholangiocytes using the QIAamp DNA Micro kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. For samples with low DNA yields (which included all FACS obtained samples), whole-genome amplification (WGA) using the Qiagen REPLI-g Mini kit (Qiagen) was performed.
Genetic analysis by microarray and genotyping
We assessed CNVs, LOH regions, and regions of homozygosity using genome-wide high-resolution cytogenetic array analysis (CytoScan HD, Affymetrix). We screened whole-blood PLD patient DNA for germline variants in PKD1 (NG_008617.1, NM_001009944.2), PKD2 (NG_008604.1, NM_000297.3), PRKCSH (NG_009300.1, NM_002743.3), SEC63 (NG_008270.1, NM_007214.4), and LRP5 (NG_015835.1, NM_002335.3) using direct sequencing as described previously.11 Briefly, we isolated DNA from whole blood using the PureGene DNA isolation kit (Gentra Systems/Qiagen, Minneapolis, MN, USA) or High Pure Polymerase chain reaction (PCR) template preparation kit (Roche, Mannheim, Germany), and stored it at 4 °C. PCR amplified PKD2, PRKCSH, SEC63, and LRP5, exons and flanking intronic sequences with specific primers (Supplementary Table S1). Screening for germline variants in PKD1 (NG_008617.1, NM_001009944.2) was carried out by a method adapted from Tan et al.20 In short, primer sequences (Supplementary Table S1) for long range PCR were chosen on specific regions of the PKD1 gene preventing the amplification of the known duplications of the first 33 exons at the proximal side of the gene. PCR reactions were performed according to the manufacturer’s manual using FastStart Taq DNA Polymerase System supplemented with GC-RICH solution (Roche) for exon 1 or GeneAmp High Fidelity PCR System (Life technologies, Carlsbad, CA, USA) supplemented with 5% DMSO for all other fragments. Annealing temperatures during PCR were carefully selected for each amplicon to amplify the desired regions. After purification of PCR amplicons from gel using the QIAEXII Gel Extraction Kit (Qiagen), a total of 500 ng of equimolar amounts of the PCR amplicons of each sample were sequenced in a single run using Ion Torrent Next-Generation Sequencing (Life Technologies).
All cysts from patients with PRKCSH variants were analyzed for LOH using direct sequencing of this variant. We screened the entire coding region of BUB1 Mitotic Checkpoint Serine/Threonine Kinase (BUB1) (NG_012048.1, NM_004336.4) in a cohort of unrelated patients with ADPLD (n=100) that met the Reynolds’ criteria.21, 22 We performed high-resolution melting curves (RotorGene-Q; Qiagen) to reveal differences in melting curve shape that correlate to BUB1 genotype variants and validated these findings by Sanger sequencing. Adenylate Cyclase 1 (ADCY1) (NG_034198.1, NM_021116.2), Insulin-Like Growth Factor Binding Protein (IGFBP) 1 (NC_000007.14, NM_000596.2), and IGBPF3 (NG_011508.1, NM_001013398.1) were analyzed in a patient with cyst chromosome 7 copy-number loss. Exons and flanking intronic sequences were amplified using PCR with specific primers (Supplementary Table S1). We purified PCR amplicons from gel using the QIAEXII Gel Extraction Kit (Qiagen) and sequenced them with the BigDye terminator kit (Applied Biosystems, Foster City, CA, USA) and ABI3730, ABI310, or ABI3100 Genetic Analyzers (Applied Biosystems, Boston, MA, USA), or (from 2014 onwards) Ion Torrent sequencing (Life Technologies).
Data analysis
We considered large regions of homozygosity on the autosomes (>3.0 Mb) and large CNVs (>1.0 Mb) for further analysis. For samples not derived by FACS/WGA, we placed no limits for size of LOH or CNVs around known cyst genes. We only assessed X chromosomes for whole-chromosome abnormalities. For selected cases, we compared the array results derived from cyst cell DNA (somatic) and germline DNA (genomic) to ensure identification of somatic events. We selected cases for germline analysis based on FACS/WGA origin of cyst DNA or presence of non-mosaic >1.0 Mb CNVs. We used individual CytoScan HD data chromosome analysis suite (CHAS, Affymetrix) V2.1. For analysis involving multiple SNP array data, we used Nexus Copy Number (Biodiscovery, El Segundo, CA, USA) V6.0.
SNP array data
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE78808 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=kjipyeauhnwbzqv&acc=GSE78808). Described variants have also been deposited at NCBI’s database of genomic structural variations (Wills et al, 2016) and are accessible through dbVar (http://www.ncbi.nlm.nih.gov/dbvar) accession number nstd125.
Results
Patient characteristics
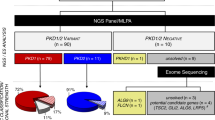
We included DNA from 24 liver cysts from 23 patients for Affymetrix CytoScan HD SNP array analysis in this study (Figure 1, Table 1; Supplementary Figure S1 for inclusion flow chart), together with 23 cysts from six previously described patients (Supplementary Table S2; Supplementary Figure S2). In the new cohort, all patients except one were females, and had single or multiple liver cysts. All patients had severe symptoms and the mean age was 54 (range 42–83) years. Seventeen patients had ADPLD, three had ADPKD, and three had solitary or sporadic cysts. Six carried PRKCSH variants, one carried a variant in SEC63, and two had PKD1 variants.

Overview of abnormal regions found in this study by SNP array.
Germline variants
In the total cohort, three patients displayed germline abnormalities detectable by our criteria outside of PRKCSH, SEC63, LRP5, PKD1, and PKD2 genomic regions. We found a heterozygous 3.0 Mb 2q13 complex rearrangement in germline and cyst DNA of a PRKCSH variant carrier (patient 8; Figure 2). This rearrangement constituted a copy-number gain as well as a copy-number loss, and contained the genes BUB1, ACOXL, BCL2L1, ANAPC1, MERTK, TMEM87B, FBLN7, and ZC3H8* (Figure 3). Peripheral blood and cyst DNA of another patient (#1) displayed a large copy-number loss (12.7 Mb) of chromosome 3p containing Wnt signaling effector catenin (cadherin-associated protein) beta 1 (CTNNB1), among others (Figure 4). We additionally identified a large copy-number loss (2.9 Mb) in patient #1, which contained the genes SYK, NFIL3, and ROR2**. This CNV in chromosome 9 occurs with low frequency in a normal, healthy cohort. A third ADPLD patient carried a gain of the entire X chromosome (Triple-X) in her germline and somatic DNA (patient 18; Figure 2).

Upper panel: 2q13 complex rearrangement present in genomic blood DNA of PRKCSH variant patient (#8). Lower panel: presence of three X chromosomes in genomic blood DNA of blood of another PRKCSH variant patient (#19).

BUB1 is present at the centromeric part of the 2q13 loss region.

Left: CNN LOH on chromosome 3p of patient #18, 19, and 1, from above to below in that order. Right upper panel: mosaic whole-chromosome 7 gain in an ADPLD patient (#11) with unknown variant, chromosome 7p shown. Right lower panel: 1.7 Mb loss on chromosome 7p of a PRKCSH patient cyst (#8.2).
* Acyl-CoA Oxidase-Like (ACOXL); BCL2-Like 1 (BCL2L1); Anaphase Promoting Complex Subunit 1 (ANAPC1); MER Proto-Oncogene, Tyrosine Kinase (MERTK); Transmembrane protein 87B (TMEM87B); Fibulin 7 (FBLN7); Zinc-Finger CCCH-Type Containing 8 (ZC3H8); ** Spleen Tyrosine Kinase (SYK); Nuclear Factor, Interleukin 3 Regulated (NFIL3); Receptor Tyrosine Kinase-Like Orphan Receptor 2 (ROR2).
Germline BUB1 variants
The 2q13 rearrangement pointed to the well-known mitotic checkpoint gene BUB1.23 To test whether this gene had a more general role in ADPLD, we studied the gene in genomic DNA from 100 severely affected ADPLD patients. Using high-resolution melt curves and subsequent Sanger sequencing on abnormal melting curves, we identified one synonymous SNP (rs370559107: hg38 chr2:g.110672765C>G, silent variant) in these 100 patients. In addition, a single, nonsynonymous SNP (rs61730706; hg38 chr2:g.110667649G>A, c.677C>T, p.(Ala226Val)) was present in the DNA of one patient of this cohort, which occurs with a minor allele frequency of 0.002–0.005 in the general population. Polyphen and SIFT showed conflicting effects (SIFT: 0.28, tolerated; Polyphen (HumDiv): possibly damaging 0.749; Polyphen (HumVar) benign 0.219; Variant taster variant (P=0.617)). We did not consider this SNP as causative or disease-related, as it is normally present in the general population.
LOH surrounding PRKCSH, SEC63, LRP5, PKD1, and PKD2
To validate our approach, the SNP array cohort included seven cysts from six patients with known PRKCSH variants. The SNP arrays of five cysts of four patients revealed copy-number-neutral (CNN) LOH on chromosome 19 at the position where PRKCSH is located (Supplementary Figure S4). Sanger sequencing of additional cyst DNA from patients (#12, 17, and 19) with known PRKCSH variants revealed somatic loss of the respective wild-type alleles in 17 out of 22 cysts (Supplementary Table S3). DNA of a cyst of patient #7 contained a >30 Mb homozygous region on chromosome 16p. Although initially classified as having isolated PLD with a single renal cortical cyst, a germline PKD1 variant was found (hg38 chr16:g.2090007T>G, c.12632A>C, p.(Glu4211Ala)). No CNN LOH was found in cyst DNA of the other two ADPKD patients by SNP array, and we proceeded by directly sequencing PKD1 and PKD2. Patient #18 had a germline PKD1 variant (hg38 chr16:g.2135508G>A, c.182C>T, p.(Pro61Leu)). In the cyst, the second allele was likely affected by a somatic deletion (hg38 chr16 g.[2135508G>A(;)2090517_2090558delinsGGAC], c.[182C>T(;)12171_12212delinsGTCC], p.[Pro61Leu(;)Ser4057Argfs*87]). We detected no variants in germline or somatic DNA of the final ADPKD patient.
LOH across ADPLD genes was a frequent occurrence in somatic DNA of our total cohort, regardless of variant status of germline DNA. Approximately 40% of 34 cysts with high-quality SNP array data displayed LOH or allelic imbalance (AI) across PRKCSH, whereas >25% of cysts had PKD1 LOH or AI. PKD2, SEC63, and LRP5 all had lower percentages of LOH/AI ~15, ~15 and ~10%, respectively. Copy-number losses and gains in ADPLD occurred less frequently.
Novel, digenic abnormalities in cysts
As described above, patient #8 presented with two germline events. We identified a PRKCSH variant and a complex rearrangement resulting in BUB1 haploinsufficiency. DNA from her second cyst displayed multiple second hits when compared with peripheral blood. A 1.7 Mb copy-number loss region on chromosome 7 was present as well as CNN LOH surrounding PRKCSH on chromosome 19 (Figure 4). The loss region includes the genes ADCY1, IGFBP1, and IGFBP3, as well as the pseudogene Septin 7B2 (SEPT7B2). It was not detected in the SNP array profile of germline DNA, nor were germline variants detected in any of these genes using Sanger sequencing. Sanger sequencing also did not reveal variants in these genes in the cyst. Another PRKCSH mutant cyst displayed mosaic trisomy 7 (#11), although only present in a subset of cells. Copy-number-neutral LOH of chromosome 19 (1.2 Mb), encompassing PRKCSH, and chromosome 11 (1.3 Mb), encompassing LRP5, was present in cyst DNA of an ADPKD patient (#18, germline PKD1 variant). In this patient, an additional copy-number-neutral LOH region was detected on chromosome 3p (hg19 chr3:g.49,735,745_53,133,526), partially overlapping with a region of a PRKCSH variant carrier (#19; hg19 chr3:g.46,715,645_52,852,488). Notably, patient #1 with a larger copy-number-neutral LOH on chromosome 3p on germline and cyst DNA also had overlap (hg 19 chr 3:g.39,874,567_52,653,645). The minimal region of LOH contained >60 genes, containing multiple tumor suppressor genes (Supplementary Table S4).
Sporadic cysts
Two sporadic cysts of the same patient showed novel chromosomal LOH (patient #28; Supplementary Table S2). These cysts had been previously described without abnormalities encompassing the then known cyst genes PRKCSH, SEC63, PKD1, or PKD2. Copy-number-neutral LOH (2.0 Mb) was found around LRP5, a novel cyst gene (Supplementary Figure S5). Germline DNA revealed no variant in LRP5, and the presence of two germline heterozygous LRP5 SNPs (hg38 chr.11:g.68403545T>C, rs545382; hg38 chr.11:g.68425222G>A, rs556442) indicated that LOH was only present at a somatic level. Unfortunately, no cyst DNA remained, and it could not be confirmed whether this copy-number-neutral LOH led to loss of wild-type alleles by variant. Finally, the sporadic cyst from patient 20 displayed a 3.8 Mb mosaic copy-number gain at the telomeric part of chromosome 16p on cytoscan. The signal was relatively weak, yet may suggest presence of a cellular subpopulation of this cyst with PKD1 gain.
Discussion
Here, we show that germline and somatic abnormalities outside of known cyst genes frequently occur in PLD, and many cysts of PLD patients have a unique genetic signature. LOH in known regions was confirmed for PRKCSH (22/29) and PKD1/PKD2 (2/3) variants. In the SNP array cohort of 23 new patients (24 cysts), we detected 12 cysts with sizable copy-number losses or LOH outside of earlier identified genomic regions. In three patients, we found the presence of unique germline aberrations in PLD. In addition, a patient with a primary liver phenotype had a germline PKD1 variant. Cyst DNA showed recurrent copy-number loss on chromosome 3, with overlap between three patients. On chromosome 7, a 1.7 Mb copy-number loss and a mosaic whole-chromosome gain were found. Sporadic cysts displayed LOH around LRP5 and a mosaic gain surrounding PKD1 at a somatic level.
Although many genomic abnormalities were present beyond genomic regions known for second hits, most were unique. Chromosomal mapping suggests that each cyst follows an independent genetic pathway, similar to mosaicism observed in tissues of other somatic second-hit disorders.24, 25 This likely reflects significant heterogeneity that is at the genomic root of cyst development. It also reflects that general genomic instability presumably precedes cyst development.16, 17 BUB1 haploinsufficiency is a known driver of chromosomal instability resulting in LOH and tumorigenesis,23, 26 and a 2q13 microdeletion has previously been described.26, 27 Although we show that it is unlikely that this specific gene is generally involved in LOH in PLD, other drivers of genetic instability may be present in the disease. Wnt signaling abnormalities appear to be common in PLD. Wnt signaling abnormalities are associated with tumorigenesis in the liver and kidney, and cause aberrant cyst development in mice.28, 29 CTNNB1 deletions and APC variants are also frequently found in hepatoblastoma.30, 31 Variants in canonical Wnt signaling component LRP5 causes ADPLD in man,4 whereas the c-terminal tail of polycystin-1 might interact with beta-catenin.32 In the same vein, our finding of copy-number-neutral LOH around CTNNB1 and with copy-number loss of ROR233 in the germline of an ADPLD patient, together with copy-number-neutral LOH around LRP5 in two sporadic cysts of another patient, further implicates this pathway as crucial for cystogenesis.
Trisomy X, the final germline abnormality we found, is unlikely to be involved in cystogenesis. Although female gender is a risk factor for cyst development and complications,34 this is most likely owing to the sex hormones.35, 36 Furthermore, gene expression of triple-X cells is largely limited to one copy by X-inactivation, excluding 5–10% of genes on the X chromosome located in pseudoautosomal regions.37 We see this as a chance finding, considering its prevalence of 1 in 1000 females. On a somatic level, possible digenic or transheterozygous variants, and a PKD1 gain were present on cysts. The 1.7 Mb chromosome 7 copy-number loss of a PRKCSH variant carrier might point toward a novel transheterozygous modifier region. Haploinsufficiency of genes such as IGFBP1, IGFBP3, and ADCY1 might be relevant in cyst development, as these genes are related to the IGF and adenylyl cyclase pathway that the anti-cystogenic somatostatin analogs are involved in.38, 39, 40, 41 Surprisingly, another cyst displayed a mosaic gain over the whole of chromosome 7, indicating that overexpression of these or other genes may also be relevant for cyst development. More difficult is the recurrent, overlapping LOH region on chromosome 3 (49,735,745-52,653,645). Although two cysts display clear LOH, the third may have had a partially normal cellular subpopulation. Over 60 genes are present at this location, none of which are known cyst-related genes. This 3p21.3 region does contain a cluster of tumor suppressor genes,42 such as tumor susceptibility gene Rassf1a.43 The cluster frequently undergoes LOH in early formation of different tumors,42 including hepatocarcinomas and cholangiocarcinomas.44 The mosaic gain of PKD1 in one cyst may be consistent with overexpression of Pkd1 in mice leading to cystogenesis.45 The mosaicism of this region was relatively low however, and may represent an artifact. Given the substantial investment that went into genotyping this cohort, we express our disappointment that we found no more specific leads to clarify the origination of multiple cysts in the liver. Concluding, our chromosomal mapping indicates significant genetic heterogeneity outside of known second-hit regions of liver cysts. We identified unique cystogenic regions, as well as characteristics of general genomic instability in hepatic cyst DNA. These findings may explain the large number of APDLD cases without a known variant, as well as phenotypic dissimilarities between similar cyst germline variants.
Accession codes
References
Bae KT, Zhu F, Chapman AB et al: Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol 2006; 1: 64–69.
Pirson Y : Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 2010; 17: 173–180.
Waanders E, te Morsche RH, de Man RA, Jansen JB, Drenth JP : Extensive variantal analysis of PRKCSH and SEC63 broadens the spectrum of polycystic liver disease. Hum Mutat 2006; 27: 830.
Cnossen WR, te Morsche RH, Hoischen A et al: Whole-exome sequencing reveals LRP5 variants and canonical Wnt signaling associated with hepatic cystogenesis. Proc Natl Acad Sci USA 2014; 111: 5343–5348.
Harris PC, Torres VE : Polycystic kidney disease. Annu Rev Med 2009; 60: 321–337.
Wills ES, Roepman R, Drenth JP : Polycystic liver disease: ductal plate malformation and the primary cilium. Trends Mol Med 2014; 20: 261–270.
Fedeles SV, Gallagher AR, Somlo S : Polycystin-1: a master regulator of intersecting cystic pathways. Trends Mol Med 2014; 20: 251–260.
Fedeles SV, Tian X, Gallagher AR et al: A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat Genet 2011; 43: 639–647.
Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S : Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet 2013; 45: 1004–1012.
Janssen MJ, Salomon J, Te Morsche RH, Drenth JP : Loss of heterozygosity is present in SEC63 germline carriers with polycystic liver disease. PLoS One 2012; 7: e50324.
Janssen MJ, Waanders E, Te Morsche RH et al: Secondary, somatic variants might promote cyst formation in patients with autosomal dominant polycystic liver disease. Gastroenterology 2011; 141: e2052.
Pei Y : A "two-hit" model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7: 151–156.
Pei Y, Watnick T, He N et al: Somatic PKD2 variants in individual kidney and liver cysts support a "two-hit" model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1999; 10: 1524–1529.
Watnick T, He N, Wang K et al: Variants of PKD1 in ADPKD2 cysts suggest a pathogenic effect of trans-heterozygous variants. Nat Genet 2000; 25: 143–144.
Wu G, Tian X, Nishimura S et al: Trans-heterozygous Pkd1 and Pkd2 variants modify expression of polycystic kidney disease. Hum Mol Genet 2002; 11: 1845–1854.
Li M, Qin S, Wang L, Zhou J : Genomic instability in patients with autosomal-dominant polycystic kidney disease. J Int Med Res 2013; 41: 169–175.
Gogusev J, Murakami I, Doussau M et al: Molecular cytogenetic aberrations in autosomal dominant polycystic kidney disease tissue. J Am Soc Nephrol 2003; 14: 359–366.
Janssen MJ, Salomon J, Cnossen WR, Bergmann C, Pfundt R, Drenth JP : Somatic loss of polycystic disease genes contributes to the formation of isolated and polycystic liver cysts. Gut 2014; 64: 688–690.
Huch M, Gehart H, van Boxtel R et al: Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2014; 160: 299–312.
Tan YC, Michaeel A, Blumenfeld J et al: A novel long-range PCR sequencing method for genetic analysis of the entire PKD1 gene. J Mol Diagn 2012; 14: 305–313.
Reynolds DM, Falk CT, Li A et al: Identification of a locus for autosomal dominant polycystic liver disease, on chromosome 19p13.2-13.1. Am J Hum Genet 2000; 67: 1598–1604.
Pei Y, Obaji J, Dupuis A et al: Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol 2009; 20: 205–212.
Baker DJ, Jin F, Jeganathan KB, van Deursen JM : Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell 2009; 16: 475–486.
Lindhurst MJ, Sapp JC, Teer JK et al: A mosaic activating variant in AKT1 associated with the Proteus syndrome. N Engl J Med 2011; 365: 611–619.
Dunn JM, Phillips RA, Becker AJ, Gallie BL : Identification of germline and somatic variants affecting the retinoblastoma gene. Science 1988; 241: 1797–1800.
de Voer RM, Geurts van Kessel A, Weren RD et al: Germline variants in the spindle assembly checkpoint genes BUB1 and BUB3 are risk factors for colorectal cancer. Gastroenterology 2013; 145: 544–547.
Yu HE, Hawash K, Picker J et al: A recurrent 1.71 Mb genomic imbalance at 2q13 increases the risk of developmental delay and dysmorphism. Clin Genet 2012; 81: 257–264.
Decaens T, Godard C, de Reynies A et al: Stabilization of beta-catenin affects mouse embryonic liver growth and hepatoblast fate. Hepatology 2008; 47: 247–258.
Saadi-Kheddouci S, Berrebi D, Romagnolo B et al: Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the beta-catenin gene. Oncogene 2001; 20: 5972–5981.
Blaker H, Hofmann WJ, Rieker RJ, Penzel R, Graf M, Otto HF : Beta-catenin accumulation and variant of the CTNNB1 gene in hepatoblastoma. Genes Chromosomes Cancer 1999; 25: 399–402.
Giardiello FM, Petersen GM, Brensinger JD et al: Hepatoblastoma and APC gene variant in familial adenomatous polyposis. Gut 1996; 39: 867–869.
Lal M, Song X, Pluznick JL et al: Polycystin-1 C-terminal tail associates with beta-catenin and inhibits canonical Wnt signaling. Hum Mol Genet 2008; 17: 3105–3117.
Geng M, Cao YC, Chen YJ, Jiang H, Bi LQ, Liu XH : Loss of Wnt5a and Ror2 protein in hepatocellular carcinoma associated with poor prognosis. World J Gastroenterol 2012; 18: 1328–1338.
Gevers TJ, Drenth JP : Diagnosis and management of polycystic liver disease. Nat Rev Gastroenterol Hepatol 2013; 10: 101–108.
Sherstha R, McKinley C, Russ P et al: Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology 1997; 26: 1282–1286.
Chapman AB : Cystic disease in women: clinical characteristics and medical management. Adv Ren Replace Ther 2003; 10: 24–30.
Tartaglia NR, Howell S, Sutherland A, Wilson R, Wilson L : A review of trisomy X (47,XXX). Orphanet J Rare Dis 2010; 5: 8.
Caroli A, Perico N, Perna A et al: Effect of longacting somatostatin analogue on kidney and cyst growth in autosomal dominant polycystic kidney disease (ALADIN): a randomised, placebo-controlled, multicentre trial. Lancet 2013; 382: 1485–1495.
Hogan MC, Masyuk TV, Page LJ et al: Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol 2010; 21: 1052–1061.
Caroli A, Antiga L, Cafaro M et al: Reducing polycystic liver volume in ADPKD: effects of somatostatin analogue octreotide. Clin J Am Soc Nephrol 2010; 5: 783–789.
Gevers TJ, Drenth JP : Somatostatin analogues for treatment of polycystic liver disease. Curr Opinion Gastroenterol 2011; 27: 294–300.
Hesson LB, Cooper WN, Latif F : Evaluation of the 3p21.3 tumour-suppressor gene cluster. Oncogene 2007; 26: 7283–7301.
Tommasi S, Dammann R, Zhang Z et al: Tumor susceptibility of Rassf1a knockout mice. Cancer Res 2005; 65: 92–98.
Tischoff I, Markwarth A, Witzigmann H et al: Allele loss and epigenetic inactivation of 3p21.3 in malignant liver tumors. Int J Cancer 2005; 115: 684–689.
Thivierge C, Kurbegovic A, Couillard M, Guillaume R, Cote O, Trudel M : Overexpression of PKD1 causes polycystic kidney disease. Mol Cell Biol 2006; 26: 1538–1548.
Acknowledgements
We thank our patients and their families for their participation and providing samples. We appreciate the input of the Genomic Disorders Group and the Molecular Biology of Ciliopathies group from the Department of Human Genetics, Radboud University Medical Center. We thank Dr Erwin van Geffen, Department of Surgery, Jeroen Bosch ziekenhuis for his valuable assistance in obtaining and processing the samples and the data. This work was supported by a grant of the Institute for Genetic and Metabolic Disease (IGMD) of the Radboud University Medical Center and by a grant from the Dutch foundation of digestive diseases WO10-50 (MLDS) (both to ESW and WRC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Wills, E., Cnossen, W., Veltman, J. et al. Chromosomal abnormalities in hepatic cysts point to novel polycystic liver disease genes. Eur J Hum Genet 24, 1707–1714 (2016). https://doi.org/10.1038/ejhg.2016.97
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.97
This article is cited by
-
Genetics, pathobiology and therapeutic opportunities of polycystic liver disease
Nature Reviews Gastroenterology & Hepatology (2022)
-
Long-term outcome of incidental cystic liver tumors in the general population
Scientific Reports (2021)
-
A human multi-lineage hepatic organoid model for liver fibrosis
Nature Communications (2021)
