
Abstract
Biotinidase deficiency is a rare inherited metabolic disorder that can cause severe neurological symptoms. To prevent severe clinical presentations, it was included in the Dutch neonatal screening programme in 2007. Since then the number of cases detected has been high. This study set out to describe the incidence of the disease, the clinical and demographic characteristics of the neonates identified and the type of mutations found. In the south-western Netherlands, 304 982 neonates were screened between 2007 and 2012; and 92 were identified for further testing. Confirmatory testing revealed 6 (7%) with a profound biotinidase deficiency (<10% enzyme activity), 44 (48%) with a partial deficiency (10–30%) and 42 (46%) with normal activity (>30%). All six patients whose profound deficiency was confirmed had enzyme activities below 15% on neonatal screening. Mutation analysis was performed in 61 neonates: 5 ‘profound’, 35 ‘partial’ and 21 ‘normal’. All five ‘profound’ cases had two severe mutations. Comparison with the northern Netherlands showed that the frequency and types of mutation were representative for the Netherlands as a whole. The most common mutation detected was c.[1330G>C] (p.(Asp444His); 34%), which is considered to be mild, followed by three severe mutations c.[1368A>C], c.[1595C>T] and c.[1330G>C;511G>A]. Seven new mutations were identified. We conclude that neonatal screening for profound biotinidase produces a high number of false positives. Biotinidase deficiency was profound in less than 10% of cases identified. As biotinidase activity lay below 15% on neonatal screening in all such cases, the screening threshold might be reduced to 15%.
Similar content being viewed by others


Introduction
Biotinidase deficiency (OMIM #253260) is a rare autosomal recessive disorder of biotin metabolism.1, 2, 3, 4, 5, 6 The biotinidase gene (BTD: OMIM 609019) is located on chromosome 3q25.7 The encoded enzyme, biotinidase, is required to separate the essential vitamin biotin from either the ingested protein-bound form or biotin-dependent enzymes in the body.2, 4 Biotin (vitamin H or B8) is important for fatty-acid synthesis, amino-acid catabolism and gluconeogenesis. Its function is to act as a co-factor for the carboxylase enzymes involved in these three processes.3, 5, 6, 8, 9 Biotinidase deficiency can cause insufficient biotin to be made available for the maintenance of these processes; this can lead to acidosis or lactic acidosis, abnormal catabolism and/or hypoglycaemia. These metabolic abnormalities can in turn lead to neurological symptoms such as seizures, hearing loss, developmental delay and/or cutaneous symptoms such as rash and alopecia.2, 4, 6
Different mutations in BTD10 may result in varying levels of biotinidase activity. Patients with the lowest enzyme activities usually present with the severest symptoms. Once cerebral damage, developmental delay and/or hearing loss have occurred in patients with biotinidase deficiency; they are usually irreversible. To prevent these symptoms, timely initiation of treatment with oral free biotin is essential,6 which explains why various countries have included biotinidase deficiency in their neonatal screening programmes.11
Only patients with a profound biotinidase deficiency appear to be at risk of developing severe and irreversible symptoms, and should therefore be the main target of screening. Although screening has also been advocated for partial deficiency,12 few individuals with a partial deficiency have been known to develop symptoms. Not only were the symptoms in these cases, such as skin-lesions and hair loss, generally mild and easily treated with biotin,6, 11, 13, 14 they usually presented after an additional trigger such as infection.6, 11
Biotinidase deficiency was included in the Dutch neonatal screening programme in 2007. The aim of this study was to gain insight into the incidence of biotinidase deficiency in the Netherlands (both profound and partially deficient) and to describe this population in terms of their biotinidase mutations and clinical characteristics.
Patients and methods
Patients and data
We included all neonates from the south-western Netherlands who had screened positive for biotinidase deficiency on neonatal screening between 2007 and 2012 (biotinidase activity ≤30% until 1 July 2012 and ≤20% from 1 July 2012). Screening for biotinidase deficiency in the Netherlands is based on a single test.
Confirmatory testing, initiation of treatment with biotin and monitoring were performed at Erasmus MC University Medical Center and, until 2010, also at Leiden University Medical Center (LUMC; who outsourced the testing to the Academic Medical Center (AMC) in Amsterdam). Patients whose confirmatory enzyme activity test was 30% or lower (or ≤20% from 1 July 2012 onwards) were treated with biotin and monitored. Neonates with an activity above 30% were regarded as not being at risk and follow-up was discontinued.
For all neonates identified through screening, information on enzyme activity was collected at screening and confirmatory testing. If available, information on disease-specific mutations was also collected. Demographic and clinical characteristics were obtained from patients’ medical files (which were available for 84–86% of neonates referred), and included gender, age at screening and confirmation, gestational age, weight, complications during pregnancy, jaundice and disease-specific symptoms (such as seizures, hearing or visual problems, and skin or hair lesions).
To determine whether disease-specific mutations were not merely particular to the south-western Netherlands, we compared the mutations with those found in the country’s northern region, that is, patients seen at University Medical Center Groningen. To determine whether the frequency of biotinidase deficiency was region specific, it was compared with the frequency in the Netherlands as a whole.
Enzyme activity testing
The initial neonatal screening test, that is, assessment of biotinidase activity on dried blood spot (DBS), was performed at the screening laboratories of the Dutch National Institute of Public Health and the Environment (RIVM). The Quantase Neonatal Biotinidase Deficiency Screening kit (Bio-Rad, Veenendaal, the Netherlands) was used to measure the biotinidase activity on DBS. Neonates with biotinidase activity below 30% were referred for confirmatory testing.
Confirmatory testing was performed by specialized metabolic laboratories at the academic centres in the Netherlands. Biotinidase activity was measured in plasma using a colorimetric assay (wavelength A546) with the artificial substrate N-biotinyl-p-aminobenzoate described by Wolf et al.15
Profound biotinidase deficiency was defined as enzyme activity that was less than 10% of the mean activity in healthy individuals, and partial deficiency as enzyme activity that was 10–30%. Biotinidase activity above 30% was regarded as sufficient and thereby considered to be normal.1, 2, 6, 14, 16 These three biotinidase activity groups refer to the enzyme activity at confirmatory testing.
Biotinidase activity was reported as a percentage of the activity (in nmol/h/ml) of healthy controls. The interval between the initial neonatal DBS screening test and the confirmatory test ranged from 1 to 13 days.
DNA analyses
DNA was isolated from peripheral blood cells according to standard procedures, and PCR and Sanger sequence analysis was used to screen all coding BTD exons and exon/intron boundaries up to 20 bp intronic, according to genomic reference NG_008019.1, transcript NM_000060.3. The software program Alamut version 2.6.1 (Interactive Biosoftware, Rouen, France) was used to help with the interpretation of sequence variants (eg, splice-site prediction). Variants have been submitted to the Leiden Open Variation Database (LOVD) on BTD (https://grenada.lumc.nl/LOVD2/shared1/home.php?select_db=BTD).
DNA mutations were classified as severe if the mutation had previously been described as leading to profound enzyme activity. We classified as mild mutations that had been described only in patients with a partial deficiency and not in patients with a profound deficiency. If insufficient information was available on the effect of the mutation/variation, a mutation was presented as an ‘unclassified variant’.
Results
Numbers of neonates screened, detected and diagnosed
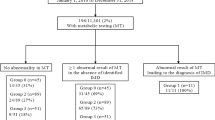
In the south-western Netherlands, 304 982 neonates were screened between 2007 and 2012. Of these, 92 were identified by neonatal DBS screening for biotinidase deficiency. Confirmatory testing revealed a biotinidase deficiency in 50 infants, representing an incidence of 1 in 6100 in the south-western region. In the Netherlands as a whole, 261 of 913 965 neonates were identified by DBS between 2008 and 2012 and biotinidase deficiency was confirmed in 111 cases, representing an incidence of 1 in 8 233. The similarity between the figures suggests that the incidence of biotinidase deficiency in the southwest is not particular to this region. (NB: the national figures excluded 2007, as data on confirmed cases were not reported in 2007). The number of cases and false positives identified were equally distributed across the seasons.
Of the 92 neonates referred in the south-western region, 6 (6.5%) were confirmed as having a profound deficiency (<10% activity) and 44 (47.8%) as having a partial deficiency (10–30% activity); 42 (45.7%) were ‘normal’ (enzyme activity >30%) and were considered to be false positives (Table 1 and Figure 1). No false negatives were identified in the Netherlands.

Biotinidase activity (% of normal) at neonatal DBS screening and at confirmation (92 neonates, south-western Netherlands, 2007–2012). Normal=biotinidase activity >30%; partial=10–30%; profound=<10% biotinidase activity on confirmatory testing. Screening test refers to neonatal DBS screening; confirmatory test refers to confirmatory test by the academic centre after referral of the neonate identified in DBS screening.
Comparison of enzyme activity at confirmatory testing and screening
Figure 1 shows the enzyme activity measured at confirmatory testing and that measured on neonatal DBS screening of the 92 neonates identified. Biotinidase activity in all six neonates with a profound biotinidase deficiency on confirmatory testing lay below 15% on DBS. All other neonates whose enzyme activity lay between 15 and 30% on DBS (30% is the cutoff value) were diagnosed as partially deficient or ‘normal’ on confirmatory testing. There was one exception, where the confirmatory test revealed 75% biotinidase activity, while the initial neonatal DBS screening result was 7%. This prompted us to ask for a retest of the DBS test in this particular case. As the retest showed normal activity (>30%), it confirmed that the initial DBS test result had been false.
Characteristics
Table 1 shows the characteristics of the neonates identified through neonatal DBS screening for each of the three biotinidase activity groups (profound, partial, normal). Although more males (n=27) than females (n=12) had a partial biotinidase deficiency, the gender distribution was not significantly different across the three groups. Age at screening and age at confirmatory testing were similar for the three groups. Only two neonates were screened outside the recommended neonatal DBS screening period of 3–7 days after birth.17
Premature births, being small for gestational age, or complications during pregnancy were observed only in the ‘partial’ and ‘normal’ groups. Jaundice was found in all three biotinidase activity groups. Eczema and skin rashes were reported in one-third of profound, one-third of partially deficient cases and in 10% of neonates with normal activity. These differences were not statistically significant.
All patients, profound and partially deficient, are treated with biotin. At the time of the referral from the neonatal screening program only mild and non-specific skin rashes were reported in 17 of the neonates. These reports were equally distributed across deficient and normal neonates. No seizures or other potential symptoms related to the disease were reported, either at the time of diagnosis or thereafter.
DNA mutations
Mutation analysis was performed for 61 of the 92 neonates (67%) from the south-western Netherlands. Table 2 shows the mutations identified and the enzyme activity that was measured at confirmation and screening. The most common mutation was c.[1330G>C] (p.(Asp444His)), which was found on 41 alleles (34%) in 53 neonates (64%) and is considered to be mild. The next most common mutations were c.[1368A>C] (p.(Gln456His), 15%), c.[1595C>T] (p.(Thr532Met), 7%) and the combination of c.[1330G>C] with c.[511G>A] (7%) on one allele. These have all been classified as severe. The most common mutations found in the south-western Netherlands were also found in the northern region of the country, which we used as comparison to evaluate potential regional differences (Supplementary Table).
Seven new mutations were detected (Table 2, bold): c.[1090dupA], c.[459+4A>G], c.[1414G>A], c.[143G>A], c.[1525C>T], the combination c.[261C>T;895G>C] and, in the northern region, the combination c.[1330G>C;1264dupC] (Supplementary Table). The profound biotinidase deficiency of patient 61 in Table 2 (second mutation c.[1330G>C;511G>A]) suggests that the newly identified c.[1414G>A] mutation can be classified as a severe mutation. The mutations c.[1525C>T] (patient 55), c.[459+4A>G] (patient 49), c.[143G>A] (patient 29) and c.[261C>T;895G>C] (patient 19) are most likely mild.
Four of the five (80%) ‘profound’ cases for whom DNA was available had two known severe mutations; the fifth had one known severe mutation in combination with the new severe mutation described above. The vast majority of partially deficient patients had one severe mutation combined with a mild mutation on the second allele (86%, 30/35), the most common combination being c.[1330G>C] (mild) with c.[1368A>C] (severe, 12/35). One patient had a combination of two previously described mild mutations (c.[1330G>C] with c.[645C>T], 20% enzyme activity at confirmation).
In the group with normal activity, seven neonates had no mutations at all (35%), seven had only one mutation (35%), and another six had two mutations (30%), at least one of which was mild. One patient had two mutations previously classified as severe (c.[133G>A] and c.[1368A>C]) while the biotinidase activity was 44% on confirmation and 26% on neonatal DBS screening.
Discussion
In 2007, neonatal DBS screening was introduced for biotinidase deficiency in the Netherlands. Since then, a large number of neonates have been identified. Our study shows that most of these were false positives or partially deficient, thereby imposing an unnecessarily large burden on parents and the healthcare system. Only 7% of those identified by screening were diagnosed with a profound biotinidase deficiency.
When biotinidase deficiency was included in the Dutch screening programme, the worldwide survey published by Wolf et al.6, 18, 19 reported an incidence of 1:61 000, including partially deficient cases. This was used to predict the outcome of a Dutch screening programme.17 More recent publications, however, indicate that the incidence of biotinidase deficiency ranges from 1:4500 to 1:62 500 in countries that screen for this disorder (eg, Brazil, the USA, Belgium, Germany and Greece).16, 20, 21, 22, 23, 24, 25, 26, 27, 28 Our study shows that, in the Netherlands, 1 in 6100 to 1 in 8200 neonates had partial or profound biotinidase deficiency.17 Only 7% of these (~1:50 000) neonates had a profound deficiency, compared with 1:137 401 reported by Wolf. The large difference between the worldwide survey and the incidence found in our study and others16, 20, 21, 22, 23, 24, 25, 26, 27, 28 is likely due to considerable variation in the occurrence of this disorder across countries. Therefore, it is important for each country to study the incidence of biotinidase deficiency. Also, some programmes included in the survey by Wolf and co-worker18, 19 reported that they may have underestimated the incidence.
To identify possible factors associated with false-positive results, we compared the neonates’ demographic and clinical characteristics, but found no evidence for such factors in our data. Characteristics were similar between ‘profoundly deficient’, ‘partially deficient’ and ‘normal’ neonates. It has been suggested that the result of the biotinidase activity test may be negatively influenced by factors such as jaundice, and that the test should be repeated after this symptom has disappeared.20, 24 As the only information available to us was the status at confirmatory testing, our data did not allow us to assess whether jaundice affected the DBS results. Seasonal differences were not detected, suggesting that the potential effect of summer heat on the DBS samples does not explain false positives either.
Similarly, maturation of the enzyme after birth has been described as causing false-positive screening results, which may be enhanced in some premature neonates by a temporary drop in enzyme activity that is lowest at days 4–6.29 Such false-positive referrals might be prevented by a second test. In our study about 10% of false-positive neonates were prematurely born and their referral might have been prevented by a second test. On the other hand, a second test would introduce a waiting period (up to a month, until the enzyme has matured), which would cause a delay in timely diagnosis and start of therapy in the truly profound patients.
In a large number of cases, the phenotype correlated well with the mutations found. Most ‘profound’ cases had two severe mutations, most ‘partial’ cases had at least one mild mutation, and ‘normal’ neonates either had one mutation or none – in other words, they were carriers or truly ‘normal’. Some ‘normal’ neonates had one or two mild mutations.
One ‘normal’ neonate was found with two presumed severe mutations (c.[133G>A] and c.[1368A>C]), which, surprisingly, yielded an enzyme activity of 44% on confirmation. Previously, c.[133G>A] was described in a carrier for biotinidase deficiency and categorized as profound.16 Biotinidase activity levels for this carrier had a very wide range, with a maximum of 39% (reported activities: 3.2, 0.9 and 0.1 nmol/min/ ml serum; normal activity: 4.4–12)16 On this basis, we conclude that this mutation is mild rather than severe.
Seven new mutations were identified: c.[1090dupA], c.[459+4A>G], c.[1414G>A], c.[143G>A], c.[1525C>T], and two combinations: c.[261C>T;895G>C] and c.[1330G>C;1264dupC]. It is likely that c.[1414G>A] is a severe mutation: in one patient, who carried the known severe mutation c.[1330G>C;511G>A] on the other allele, it led to 0% biotinidase activity. The other six new mutations were all found in partially deficient or ‘normal’ neonates. As c.[1525C>T], c.[459+4A>G], c.[143G>A], c.[261C>T;895G>C] were found in combination with a severe mutation on the second allele, we deduced that these are relatively mild mutations. In vitro mutagenesis may be needed to provide proof. The most common disease-specific mutations found in our study were c.[1330G>C] (mild), c.[1368A>C] (severe), c.[1595C>T] (severe) and the combination c.[1330G>C;511G>A] (severe). These findings are consistent with other studies performed in the US and Brazil.3, 6, 14, 20, 30, 31
Implications for screening
The standard DBS screening cutoff level of 30% biotinidase activity produces a large number of false positives and partially deficient cases. Our results indicate that, at a cutoff level of 15%, all the profound cases would be identified, and the referral of false-positive neonates and partially deficient patients would be prevented. Prior to this study it was unclear, how far the cutoff value could be lowered without missing any profoundly deficient patients in the Netherlands. In July 2012, the Dutch cutoff value for biotinidase screening was reduced to 20%, as a first attempt to decrease the number of unwanted referrals. While referral was indeed reduced, some false positives remain, including some partially deficient patients who have consequently been labelled as ‘diseased’ and treated with biotin.
Strengths and limitations
Although our study focused on the south-western Netherlands, the incidence and type of mutations did not seem region specific in the way that was found previously for medium chain acyl co-enzyme-A dehydrogenase deficiency (MCAD) in the Netherlands.32 It is likely that our conclusions can be extrapolated to the country as a whole.
As all patients, including partially deficient patients, were treated with biotin, it was not possible to investigate whether they developed symptoms. Further research is needed to identify which patients are at risk of developing symptoms, and whether this is related mainly to the residual enzyme activity, to the mutations the patient carries, or both.
Conclusions
In the Netherlands 1 in 8200 neonates had partial or profound biotinidase deficiency during our study period. Only 7% of those referred had a profound deficiency. The inclusion of biotinidase deficiency in the Dutch neonatal screening programme led to the detection of a large number of false positives and a large number of partially deficient cases. Lowering the screening cutoff to 15% would prevent all these unwanted referrals, while detecting all neonates with a profound deficiency who are at risk of developing severe symptoms.
References
Wolf B: Biotinidase deficiency. In Pagon RA, Adam MP, Ardinger HH et al: (eds), GeneReviews [Internet]. Seattle, WA, USA: University of Washington, 2000 (updated 5 December 2013)..
Li H, Spencer L, Nahhas F et al: Novel mutations causing biotinidase deficiency in individuals identified by newborn screening in Michigan including an unique intronic mutation that alters mRNA expression of the biotinidase gene. Mol Genet Metab 2014; 112: 242–246.
Norrgard KJ, Pomponio RJ, Hymes J, Wolf B : Mutations causing profound biotinidase deficiency in children ascertained by newborn screening in the United States occur at different frequencies than in symptomatic children. Pediatr Res 1999; 46: 20–27.
Wolf B, Jensen K, Huner G et al: Seventeen novel mutations that cause profound biotinidase deficiency. Mol Genet Metab 2002; 77: 108–111.
Pomponio RJ, Hymes J, Reynolds TR et al: Mutations in the human biotinidase gene that cause profound biotinidase deficiency in symptomatic children: molecular, biochemical, and clinical analysis. Pediatr Res 1997; 42: 840–848.
Wolf B : Biotinidase deficiency: "if you have to have an inherited metabolic disease, this is the one to have". Genet Med 2012; 14: 565–575.
Cole H, Weremowicz S, Morton CC, Wolf B : Localization of serum biotinidase (Btd) to human-chromosome-3 in band-P25. Genomics 1994; 22: 662–663.
Pomponio RJ, Reynolds TR, Mandel H et al: Profound biotinidase deficiency caused by a point mutation that creates a downstream cryptic 3' splice acceptor site within an exon of the human biotinidase gene. Hum Mol Genet 1997; 6: 739–745.
Hymes J, Stanley CM, Wolf B : Mutations in BTD causing biotinidase deficiency. Hum Mutat 2001; 18: 375–381.
Procter M, Wolf B, Crockett DK, Mao R : The Biotinidase Gene Variants Registry: a Paradigm Public Database. G3 2013; 3: 727–731.
Jay AM, Conway RL, Feldman GL, Nahhas F, Spencer L, Wolf B : Outcomes of individuals with profound and partial biotinidase deficiency ascertained by newborn screening in Michigan over 25 years. Genet Med 2015; 17: 205–209.
Wolf B : Why screen newborns for profound and partial biotinidase deficiency? Mol Genet Metab 2015; 114: 382–387.
McVoy JR, Levy HL, Lawler M et al: Partial biotinidase deficiency: clinical and biochemical features. J Pediatr 1990; 116: 78–83.
Swango KL, Demirkol M, Huner G et al: Partial biotinidase deficiency is usually due to the D444H mutation in the biotinidase gene. Hum Genet 1998; 102: 571–575.
Wolf B, Grier RE, Allen RJ, Goodman SI, Kien CL : Biotinidase deficiency: the enzymatic defect in late-onset multiple carboxylase deficiency. Clin Chim Acta 1983; 131: 273–281.
Neto EC, Schulte J, Rubim R et al: Newborn screening for biotinidase deficiency in Brazil: biochemical and molecular characterizations. Braz J Med Biol Res 2004; 37: 295–299.
Rijpstra A, van der Ploeg CPB, Verkerk PH : Monitor en evaluatie van de neonatale hielprikscreening bij kinderen geboren in 2012. TNO-rapport, 2014.
Wolf B : Worldwide survey of neonatal screening for biotinidase deficiency. J Inherit Metab Dis 1991; 14: 923–927.
Wolf B, Heard GS : Screening for biotinidase deficiency in newborns – worldwide experience. Pediatrics 1990; 85: 512–517.
Borsatto T, Sperb-Ludwig F, Pinto LLC et al: Biotinidase deficiency: clinical and genetic studies of 38 Brazilian patients. BMC Med Genet 2014; 15: 96.
Milankovics I, Nemeth K, Somogyi C, Schuler A, Fekete G : High frequencies of biotinidase (BTD) gene mutations in the Hungarian population. J Inherit Metab Dis 2010; 33: S289–S292.
Cowan TM, Kazerouni NN, Dharajiya N et al: Increased incidence of profound biotinidase deficiency among Hispanic newborns in California. Mol Genet Metab 2012; 106: 485–487.
Vallejo-Torres L, Castilla I, Couce ML et al: Cost-effectiveness analysis of a national newborn screening program for biotinidase deficiency. Pediatrics 2015; 136: E424–E432.
Schulpis KH, Gavrili S, Tjamouranis J, Karikas GA, Kapiki A, Costalos C : The effect of neonatal jaundice on biotinidase activity. Early Hum Dev 2003; 72: 15–24.
Karaca M, Özgül RK, Ünal Ö et al: Detection of biotinidase gene mutations in Turkish patients ascertained by newborn and family screening. Eur J Pediatr 2015; 174: 1077–1084.
Thodi G, Schulpis KH, Molou E et al: High incidence of partial biotinidase deficiency cases in newborns of Greek origin. Gene 2013; 524: 361–362.
Küry S, Ramaekers V, Bezieau S, Wolf B : Clinical utility gene card for: Biotinidase deficiency. Eur J Hum Genet 2012, e-pub ahead of print 29 February 2012 doi:10.1038/ejhg.2012.28.
Gannavarapu S, Prasad C, DiRaimo J et al: Biotinidase deficiency: spectrum of molecular, enzymatic and clinical information from newborn screening Ontario, Canada (2007-2014). Mol Genet Metab 2015; 116: 146–151.
Suormala T, Wick H, Baumgartner ER : Low biotinidase activity in plasma of some preterm infants: possible source of false-positive screening results. Eur J Pediatr 1988; 147: 478–480.
Norrgard KJ, Pomponio RJ, Swango KL et al: Mutation (Q456H) is the most common cause of profound biotinidase deficiency in children ascertained by newborn screening in the United States. Biochem Mol Med 1997; 61: 22–27.
Pomponio RJ, Coskun T, Demirkol M et al: Novel mutations cause biotinidase deficiency in Turkish children. J Inherit Metab Dis 2000; 23: 120–128.
Derks TG, Duran M, Waterham HR, Reijngoud DJ, Ten Kate LP, Smit GP : The difference between observed and expected prevalence of MCAD deficiency in The Netherlands: a genetic epidemiological study. Eur J Hum Genet 2005; 13: 947–952.
Acknowledgements
The National Advisory Board Neonatal Screening for Metabolic Diseases (ANS-MZ). We thank Mrs IH van Veen-Hof for technical assistance.
Author contributions
MW conceived the study, and RW, MK, MW, JS and AvdP participated in its design. RW, MW, WO and KN collected clinical information. WO, FV and KN were responsible for enzyme activity testing and interpretation. EK was responsible for the processing and interpretation of the DBS screening results. RvM and JS were responsible for the molecular analysis and for interpretation of the genetic variants. KN was responsible for isolation and shipment of the materials of the UMCG cohort. FvS had oversight of the UMCG cohort and contributed discussion points around the screening debate. RW, MK, AvdP and MW were involved in the interpretation of all results. RW and MK drafted the manuscript. MK, MW and AvdP oversaw the stages of revision and editing. MW takes responsibility for the content of the manuscript, including the data and analysis. All authors helped to interpret the results and read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Wiltink, R., Kruijshaar, M., van Minkelen, R. et al. Neonatal screening for profound biotinidase deficiency in the Netherlands: consequences and considerations. Eur J Hum Genet 24, 1424–1429 (2016). https://doi.org/10.1038/ejhg.2016.65
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.65
