
Abstract
The population of the province of Newfoundland and Labrador (NL) has been a resource for genetic studies because of its historical isolation and increased prevalence of several monogenic disorders. Controversy remains regarding the genetic substructure and the extent of genetic homogeneity, which have implications for disease gene mapping. Population substructure has been reported from other isolated populations such as Iceland, Finland and Sardinia. We undertook this study to further our understanding of the genetic architecture of the NL population. We enrolled 494 individuals randomly selected from NL. Genome-wide SNP data were analyzed together with that from 14 other populations including HapMap3, Ireland, Britain and Native American samples from the Human Genome Diversity Project. Using multidimensional scaling and admixture analysis, we observed that the genetic structure of the NL population resembles that of the British population but can be divided into three clusters that correspond to religious/ethnic origins: Protestant English, Roman Catholic Irish and North American aboriginals. We observed reduced heterozygosity and an increased inbreeding coefficient (mean=0.005), which corresponds to that expected in the offspring of third-cousin marriages. We also found that the NL population has a significantly higher number of runs of homozygosity (ROH) and longer lengths of ROH segments. These results are consistent with our understanding of the population history and indicate that the NL population may be ideal for identifying recessive variants for complex diseases that affect populations of European origin.
Similar content being viewed by others

Introduction
Population isolates possess many advantages and are frequently used in genetic research because they are presumed to have reduced genetic diversity, along with increased prevalence for some diseases and the tendency for affected individuals to share ancestral haplotypes derived from a small number of founders.1, 2, 3 Different isolates warrant different study designs, especially for complex traits.1, 4 Understanding population history and genetic structure will help optimize genetic studies for mapping genes of complex traits.
The province of Newfoundland and Labrador (NL) – consisting of the island of Newfoundland, and mainland Labrador on the east coast of Canada (Supplementary Figure 1) – has been thought of as a genetic isolate.4 The NL population has been an important resource for the mapping of many Mendelian traits, both recessive and dominant.5, 6, 7, 8, 9 However, the genetic structure of the NL population has yet to receive a systematic examination.
NL today is home to three peoples of Aboriginal ancestry: the Inuit, the Innu and the Mi'kmaq. The Inuit were located exclusively in Labrador. Also in Labrador, the Innu are descended from Algonkian-speaking hunter-gatherers and were present at the time of European arrival. The Mi'kmaq are found on the island of Newfoundland and are descended from Algonkians from the eastern Canadian mainland. All three aboriginal populations have inter-bred, to varying degrees, with people of European origin.10
European immigration into Newfoundland – consisting primarily of Roman Catholics from Southeastern Ireland and Protestants from Southwestern England – occurred predominantly in the late eighteenth and early nineteenth centuries.10 Until very recently, it was uncommon for Catholics and Protestants to inter-marry. Most of these immigrants settled in small coastal communities called outports, which, in the absence of roads, were geographically and culturally isolated.
In the 1980s, Bear et al11, 12 identified persistent genetic isolation and elevated inbreeding coefficients in selected Newfoundland outports. Martin et al13 examined the population structure of 10 Newfoundland outports and found significant genetic differences, which could be explained by religious segregation and geographic isolation.
Rahman et al4 genotyped 1064 SNPs in 200 blood donors from St John’s, the capital city. They found extended runs of linkage disequilibrium (LD) with >50% of SNP pairs exhibited a D’>0.33. Using the same samples, Service et al14 found that the length of the LD map of chromosome 22 in the NL population approached that of outbred European populations. These findings contradicted the previously held belief that the NL population was characterized by extended LD.4 All these studies were on a small scale, used a limited number of genetic markers and the samples were not representative of the entire NL population.
Over the past 5–10 years, the availability of genome-wide SNP data has resulted in several studies of isolated populations.15, 16, 17, 18 These have revealed distinct genetic structures that can provide guidance on using these populations for mapping the genes for complex diseases. We decided to examine the genetic structure and founder effects within the NL population by comparing genome-wide SNP data with that from Irish, British, Native American and other HapMap3 populations. We also sought – for the first time – to document by DNA analysis the aboriginal contribution to the present NL population.
Materials and methods
Study subjects
The study was approved by the NL Health Research Ethics Board. As part of a colorectal cancer research study,19 494 unaffected individuals were recruited between 2001 and 2003 by random digital dialing across the NL to serve as controls.20 Volunteers were age matched to cancer patients but the only significant inclusion criterion was the absence of a personal history of colorectal cancer. This group should therefore be representative of the genetic make-up of the NL population whose median year of birth was 1942. Participants provided a blood sample for DNA extraction, and information on their family sufficient to construct at least a three-generation pedigree. Pedigrees were expanded as far as possible to earlier generations by reference to publically available genealogical resources, in particular the 1921, 1935 and 1945 NL census data (http://ngb.chebucto.org/). Fifty-two individuals having fewer than two generations of NL ancestry were excluded from the analysis.
Ancestral origins of the Newfoundland cohort
By reference to public records it was possible, in most cases, to determine the historic religious affiliation of each family. Where appropriate, ‘Roman Catholic’ was used as a proxy for ancestors of Irish origin and ‘Protestant’ was used as a proxy for those of English origin. Each participant completed a questionnaire that provided information on personal, parental and grand-parental ethnicity. This was used to help identify families with aboriginal ancestry. Following the initial multidimensional scaling (MDS) analysis, the ancestry of population ‘outliers’ was further investigated. The extensive public list of names on the Qalipu Mi’kmaq First Nation Band Order (http://laws-lois.justice.gc.ca/PDF/SOR-2011-180.pdf), together with genealogical methods, were used to confirm ancestral connection to aboriginal populations.
Reference populations include Irish, British, Native American and HapMap3 populations. Irish samples (IRE, n=211) were healthy controls from a study of Amyotrophic Lateral Sclerosis.21 They were unrelated individuals with self-reported Irish ethnicity for at least three generations. British samples were derived from the 1958 British Birth Cohort (BBC).22 This cohort (n=2878) consists of people born in England, Scotland or Wales in a single week of 1958 and was a representative sample of the British population at the time, including immigrant families. To balance sample sizes, 450 samples were randomly chosen for our analysis. Native American samples (NAS) were from the Human Genome Diversity Project (HGDP).23 The NAS included 108 Native American samples collected from Colombia (Colombians), Brazil (Karitiana and Surui) and Mexico (Pima and Maya). Hapmap3 samples24 consist of 11 outbred populations including those of African ancestry in the Southwest USA (ASW), Utah residents with Northern and Western European ancestry from the CEPH collection (CEU), Han Chinese in Beijing, China (CHB), Chinese in Metropolitan Denver, Colorado (CHD), Gujarati Indians in Houston, Texas (GIH), Japanese in Tokyo, Japan (JPT), Luhya in Webuye, Kenya (LWK), those of Mexican ancestry in Los Angeles, California (MEX), Maasai in Kinyawa, Kenya (MKK), Tuscans in Italy (TSI), and the Yoruba of Ibadan, Nigeria (YRI).
Genotyping
DNA extracted from the NL blood samples was genotyped using an Affymetrix Axiom Genome-Wide Array (Santa Clara, CA, USA), which contains 1.2 million SNPs. IRE samples were genotyped on Illumina Infinium II 550 K SNP chips, which contain 561 466 SNPs selected from the HapMap Project (Illumina, San Diego, CA, USA).21 The BBC samples were genotyped on the Illumina 1.2 M chip.22 The NAS samples were genotyped on Illumina 650Y arrays, which contain 660 918 markers.23 Hapmap3 samples were genotyped on both Affymetrix Human SNP array 6.0 and Illumina Human 1M-single Beadchip.25 The consensus genotype data were directly downloaded from the International HapMap Project website (http://hapmap.ncbi.nlm.nih.gov).
The genotype data of the NL samples are available at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74392 with accession number GSE74392. Religion/ethnicity data of the NL samples are available at request to the author (guangju.zhai@med.mun.ca) subject to an appropriate ethics approval.
Data analysis
All genotype data sets were subject to a series of quality control steps, which were done in each population separately. Sex consistency was checked based on genotype data on chromosome X and self-reported sex. The rate of missing data was examined at both the individual level and the SNP level. Individuals with >3% missing SNP data were excluded, as were SNPs that were not successfully typed in >5% of participants. SNPs with minor allele frequencies <5% in any population were removed, as were all SNPs with P<10−4 for the Hardy–Weinberg equilibrium test. After the quality control steps, 174 754 autosomal SNPs common to all populations were used in this study, unless otherwise indicated.
Population structure
Three MDS analyses were carried out using PLINK.26 The first included all 14 populations; the second included the NL, IRE, BBC and NAS data; the third included only the NL, IRE and BBC data. We visualized these data using two-dimensional plots created by the ggplot227 package in R (The R Foundation, Vienna, Austria). In addition, haplotype-based analysis was carried out on the data of the NL, IRE, BBC and NAS using fineStructure28 to confirm the MDS analysis results and to identify more detailed patterns of the NL population.
Ancestral clustering
The program ADMIXTURE29 was used to predict individual ancestry admixture proportions for a range of assumed numbers of distinct populations (K=2–5), and the results were visualized using the barplot package in R. F4 ratio estimation30 was used to estimate the mixing proportion of the ancestral population.
Genetic diversity
The heterozygote rate was calculated using Haploview31 and inbreeding coefficients were calculated as F=(He−Ho)/He2 where He is the expected heterozygote rate from the Hardy–Weinberg equation and Ho is the observed heterozygote rate. Runs of homozygosity (ROH) were identified using PLINK. The following parameters were applied for calling ROH: 5-Mb window size, a minimum of 50 SNPs per window and allowing one heterozygous and five missing calls per window. In addition, we required a minimum ROH length of 1 Mb and a minimum of 100 contiguous homozygous SNPs.
Linkage disequilibrium
Pairwise D’ and r2 within each 500-kb region were calculated within populations using Haploview.31 This was done on the 86 individuals randomly selected from each population. ALDER program32 was used to compute weighted LD curves to infer admixture parameters including dates, mixture proportions and phylogeny.
Results
Analysis of population structure
Of the 494 NL individuals originally selected, the 442 (89.5%) used in the genetic analyses had their ancestry in NL traced for at least three generations, typically for many more. MDS analysis of all study populations (Supplementary Figure 2) shows that the NL samples predominantly cluster with the CEU, BBC and IRE populations (most of the CEU, BBC and IRE symbols are covered by the NL symbols), with some outliers trending toward the MEX, GIH, NAS and Asian populations. Of note, the NAS samples were clustered further away from the MEX samples to Asian populations, which is consistent with the population history.33
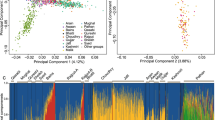
The second MDS analysis shown in Figure 1 was limited to data from the NL, IRE, BBC and NAS populations. Majority of the NL samples overlapped with the IRE and BBC samples, with few outliers trending toward to the NAS samples. These outliers were aboriginals. Noticeably, the five individual Native American populations were well separated. fineStructure analysis showed a similar pattern but also demonstrated a subtle shifting of the NL population toward to the NAS population (Supplementary Figure 3). The Native American populations were also well separated from European populations as expected in the fineStructure analysis. The NL aboriginal samples were close to the NAS, particularly the Colombian samples. However, three individual Native American populations (Karitiana, Pima and Surui) were clustered together with unknown reasons in the fineStructure analysis compared with the results from the MDS analysis.

MDS analysis of the BBC, IRE, NAS and NL populations.
The third MDS analysis shown in Figure 2 was limited to data from the NL, IRE and BBC populations. A small proportion of the NL population overlapped the IRE samples. A large proportion of the NL samples overlapped the BBC samples, with two groups of outliers trending away from the BBC cluster.

MDS analysis of the BBC, IRE and NL populations.
The overlap between the IRE and NL populations is smaller than expected given the significant contribution of Irish to the founder population of NL. We considered the possibility that the use of different genotyping platforms in different populations may have affected our MDS analyses. To test this hypothesis, we added to our analysis additional SNP data from 200 NL colorectal cancer patients who had been genotyped on both Illumina and Affymetrix platforms. We found excellent concordance between the data from the two platforms indicating that the use of different genotyping platforms was not an issue (Supplementary Figure 4).
Figure 3a uses the same three-population MDS data as in Figure 2 but, for clarity, only the NL samples are plotted, together with annotation of their religious/ethnic status (see Table 1). Significantly, this analysis was able to clearly separate Protestant (English) from Roman Catholic (Irish) families. Those individuals with documented mixed Protestant/Catholic ancestry map between the two main clusters. Perhaps surprisingly, aboriginal samples overlapped the other NL samples in this analysis. Two groups of Protestant outliers can also be recognized. Those in Group 1 all originate from one area of the province (Conception Bay), whereas those in Group 2 originate from the South and Southwest coasts of the island.

MDS analysis of the NL samples. (a) Same analysis as in Figure 2, but only the NL samples are shown. Religion/ethnicity annotations: AB, some aboriginal ancestry; C, Roman Catholic; P, Protestant; P/C, mixed Protestant and Roman Catholic ancestry. (b) Same 14-population analysis used in Supplementary Figure 2, but only the NL samples are shown. Religion/ethnicity annotations as in a.
Using a similar approach, the information in Figure 3b is derived from the same 14-population MDS data as used in Supplementary Figure 2 but, for clarity, only the NL samples are plotted. Compared with Figure 3a, the inclusion of the Asian, Native American and African comparators permits the NL aboriginal samples to be clearly distinguished from other NL individuals.
Population admixture analysis
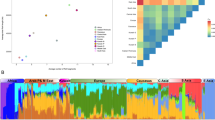
To evaluate admixture in the NL population, we used the maximum likelihood method implemented in ADMIXTURE to estimate individual admixture.29 This method allows for uncertainty in ancestral allele frequencies. We pre-defined the number of populations as K=2 to K=5, and only included the NL, IRE, BBC and NAS populations in this analysis. The results are presented in Figure 4. Using a value of K=5 appears to provide optimal information as also indicated by the lowest cross-validation (CV) error (the CV error was 0.55135, 0.55160, 0.54679 and 0.54614 for K=2, 3, 4 and 5, respectively). The NL population has a similar ancestry admixture to the IRE and BBC, except that the NL aboriginal individuals have a higher proportion of Native American ancestry. This is particularly apparent for K=2, 4 and 5 and is consistent with the pre-historic North-East Asian origin of all North American aboriginals. Using F4 ratio estimation with the phylogeny presented in Supplementary Figure 5, we found that the NL population had the proportion of British ancestry 0.59±0.22 and of Irish ancestry 0.41±0.22, but we did not find a significant proportion of Asian ancestry, most likely due to the small number of the aboriginals in our sample.

Population admixture analyses for the NL, European and Native American populations. The ancestry admixture of each individual was determined using ADMIXTURE. We performed the analyses with different numbers of theoretical ancestral populations from K=2 to K=5.
Genetic diversity
We calculated the frequencies of heterozygosity and the inbreeding coefficients for each of the European populations (IRE, BBC, CEU and TSI) and for the NL population. The calculations were made using (1) only the SNPs common to all five populations; (2) only the LD-pruned SNPs; and (3) all the available SNPs (Table 2).
When analyzing only those SNPs common to all populations, the NL population has the highest inbreeding coefficient, with an average of 0.005. This value corresponds to that expected in the offspring of third-cousin marriages. The CEU and TSI populations have negative values, suggesting the possibility of disassortative mating. The IRE and BBC populations present low inbreeding coefficients as expected for outbred populations. Furthermore, the NL population has the lowest frequency of heterozygosity, especially noticeable when all available SNPs are included in the analysis (Table 2).
The low heterozygosity and increased inbreeding coefficients complement the results of the ROH analysis. Within the autosomal genome, the average number of ROH segments per NL sample was more than double that in the IRE, BBC and TSI populations and almost triple the number in the CEU group (Figure 5a). The average length of ROH segments in the NL population was 6.73 Mb, which is more than double that in the IRE and BBC populations (Figure 5b). Of note, the NL population has many people with extreme ROH lengths (20–53 Mb), indicative of a recent common ancestor.

(a) Box-and-whisker for the average number of ROH per person in each population. (b) Box-and-whisker plot for the length of ROH, and individual outliers in each population.
LD patterns
To explore the nature and the extent of LD, we calculated D’ and r2 between all pairs of markers within 500-kb regions on each chromosome for each European population. To counter the effects of unequal sample size, we randomly selected 86 individuals (which was the total size of the TSI sample) from each population. LD patterns across the entire genome were similar for both measures in all five populations (Supplementary Figures 6a and 6b). The analysis of LD decay showed that the NL population has slightly stronger LD than BBC, CEU or TSI populations, but weaker LD than the IRE population. ALDER analysis failed to compute a two-reference weighted LD decay curve for the NL population when using both BBC and IRE as reference populations. However, when using TSI as a reference population, we found a significant one-reference weighted LD decay curve for the NL, BBC and IRE, respectively. The lower bound of the mixture fraction was estimated as 57.7±5.7, 65.3±6.5 and 58.8±16.1 generations ago for the NL, BBC and IRE, respectively, suggesting the modern NL population remains a similar LD pattern of its source populations.
Discussion
According to archeological evidence, at least three native groups inhabited NL before the arrival of Europeans: the Paleo Eskimos, the Maritime Archaic and the Beothuk.34 Of these, only the Beothuk were still extant in the seventeenth century, but the last known surviving Beothuk died in 1829. There was migration of Mi’kmaq into Southeastern Newfoundland in the eighteenth century, although it is likely that Mi’kmaq habitation of the island pre-dated the arrival of Europeans.35
The peak European immigration to the island of Newfoundland occurred in the mid-to-late eighteenth century, and included mainly Protestant settlers from the Southwest of England and Roman Catholic settlers from the Southeast of Ireland.4 Starting from about 20–25 thousand settlers in 1760, the population grew by natural expansion to about 200 000 in 1890. There was little in- or out-migration from the early nineteenth century up to fairly recent times. In the census of 1945 – approximating the median birth year of the NL cohort used in this study – the population was 321 819.36 In 1945, 33% of census respondents were reported as ‘Roman Catholic’, whereas 66% were reported as belonging to one of the Protestant faiths. In the 2011 Canadian census, the population of the province was 514 536, with 26 728 (5.2%) residing in Labrador.
Our MDS analysis is in agreement with the known population history. The NL samples cluster with European populations including BBC, IRE and CEU populations on the global population background (Supplementary Figure 2). When only the NL, BBC and IRE populations were examined, the IRE and BBC populations were separated, with some overlapping (Figure 2). A small proportion of the NL population overlapped the IRE samples, whereas a large proportion overlapped the BBC samples, although two groups of outliers trend away from the BBC cluster. The data in Figure 3 more clearly illustrate NL population substructure. The major cluster overlapping the BBC population is Protestant, whereas the cluster toward or overlapping with the IRE population represents Catholics. These observations are in agreement with the known history of the NL population. The observed genetic structure of the BBC population is in agreement with a previous report.16
The overlap between the IRE and NL populations is smaller than expected given the significant contribution of Irish to the founder population of NL. This may be due to a founder bottleneck event. The IRE samples were recruited from across Ireland, whereas the Irish people who immigrated to NL in the late eighteenth century originated mainly from a small region of Southeastern Ireland. To determine the significance of these geographical differences is difficult as, to the best of our knowledge, there has been no detailed analysis of the genetic substructure of the Irish population based on geographical origins within Ireland.
The ability of MDS analysis to separate Protestant (English) from Catholic (Irish) Newfoundlanders is strikingly illustrated in Figure 3a. The power of religion to prevent admixture between neighboring communities – over >200 years – is further illustrated by the observation that in the 407 NL families documented as having Catholic or Protestant origins, only 14 families show significant mixing of the two religions (Table 1). On the MDS analysis, these mixed families cluster between the two main religious groups. Another significant contribution to genetic substructure in the NL population is alleles derived from aboriginal ancestors. This became apparent only when including non-European samples in the MDS analysis (Figure 3b). We identified aboriginal ancestry in 20 families (4.5%). Owing to the method of ascertainment, this is likely to be a minimum estimate of the true aboriginal contribution. Only a small number of individuals provided ethnicity data on questionnaires, typically those with knowledge from a close relative. Others we designated as of aboriginal ancestry were ascertained post hoc by examining obvious outliers in the MDS analyses. We correlated family names with known aboriginal names and birthplaces, especially those published in the Qalipu Mi’kmaq First Nation Band Order (http://laws-lois.justice.gc.ca/PDF/SOR-2011-180.pdf). It is likely that had we extended this method to examine those outliers nearer or overlapping European individuals, we would have found additional evidence of aboriginal contribution, albeit at a lower admixture proportion.
Population ancestral admixture analyses showed results consistent with our population immigration history. We demonstrated that the NL population has similar ancestral proportions as other European populations, and those aboriginal individuals in the NL population tend to have a higher proportion of Native American ancestry although a significant contribution of Native American ancestry was not detected at population level with using ALDER program because of the small number of the aboriginals in our sample.
It is believed that population isolates have significantly less genetic diversity than outbred populations.1 Consequently, isolated populations are expected to exhibit increased inbreeding coefficients, increased frequencies of homozygosity and a decreased number of heterozygotes.2 The NL population has the lowest heterozygosity rate and highest inbreeding coefficient of the European populations we studied. The average inbreeding coefficient is 0.005, roughly corresponding to the children of third-cousin marriages. Analysis of ROH is a powerful method to gauge the extent of ancient kinship and recent parental relationship within a population,16 because ROH arise from a common ancestor shared by both of an individual’s parents. We found that the NL population has more than double the number of ROH across the entire genome than other European populations. We also found that the average length of ROH is more than double that in European populations. This agrees well with the observation of higher inbreeding coefficients and lower heterozygosity rates in the NL group. This explains why the NL population may be more prone to certain monogenic recessive diseases than are more outbred populations.37, 38
It had been postulated that isolated populations would exhibit high LD, but we found that the NL LD patterns and their decay with distance between SNPs are not much different from those in other European populations. In fact, at the level of D’ and r2, the populations studied seemed indistinguishable. Our findings are similar to those of a previous report in which LD was examined only within chromosome 22.14 Unlike some other isolates, the founding size of the NL population was relatively large. In such a population, there would have been relatively little opportunity for genetic drift and most people would be connected by more meiotic steps than in a rapidly growing population.39 The extent of LD in some other isolates such as the Finnish and Sardinian populations are found to be similar to those in more mixed populations like those of the United States and the United Kingdom.40, 41
In conclusion, we found genetic substructure within the NL population as a consequence of separate contributions from English, Irish and aboriginal founders. These should be taken into account when conducting genetic association studies. The NL population is also characterized by reduced heterogeneity, leading to the increased incidence of some recessive diseases. Although the population has been especially valuable for mapping rare monogenic diseases, it could also be well suited for identifying recessive variants associated with complex traits. Although cultural and environmental factors have probably reduced the signal-to-noise ratio for genetic mapping in outbred populations, the high degree of cultural and environmental homogeneity within the NL population provides a substantial mapping advantage. The reduced genetic heterogeneity will facilitate efficient genetic mapping and identification of variants – especially recessive variants – associated with complex traits.
References
Peltonen L, Palotie A, Lange K : Use of population isolates for mapping complex traits. Nat Rev Genet 2000; 1: 182–190.
Arcos-Burgos M, Muenke M : Genetics of population isolates. Clin Genet 2002; 61: 233–247.
Marjamaa A, Salomaa V, Newton-Cheh C et al: High prevalence of four long QT syndrome founder mutations in the Finnish population. Ann Med 2009; 41: 234–240.
Rahman P, Jones A, Curtis J et al: The Newfoundland population: a unique resource for genetic investigation of complex diseases. Hum Mol Genet 2003; 12 Spec No 2: R167–R172.
Warden G, Harnett D, Green J et al: A population-based study of hereditary non-polyposis colorectal cancer: evidence of pathologic and genetic heterogeneity. Clin Genet 2013; 84: 522–530.
Moore SJ, Buckley DJ, MacMillan A et al: The clinical and genetic epidemiology of neuronal ceroid lipofuscinosis in Newfoundland. Clin Genet 2008; 74: 213–222.
Merner ND, Hodgkinson KA, Haywood AF et al: Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet 2008; 82: 809–821.
Parfrey PS : Autosomal-recessive polycystic kidney disease. Kidney Int 2005; 67: 1638–1648.
Moore SJ, Green JS, Fan Y et al: Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study. Am J Med Genet A 2005; 132A: 352–360.
Mannion JJ : The Peopling of Newfoundland. Memorial University of Newfoundland: St John's, NL, Canada, 1997.
Bear JC, Nemec TF, Kennedy JC et al: Persistent genetic isolation in outport Newfoundland. Am J Med Genet 1987; 27: 807–830.
Bear JC, Nemec TF, Kennedy JC et al: Inbreeding in outport Newfoundland. Am J Med Genet 1988; 29: 649–660.
Martin LJ, Crawford MH, Koertvelyessy T, Keeping D, Collins M, Huntsman R : The population structure of ten Newfoundland outports. Hum Biol 2000; 72: 997–1016.
Service S, DeYoung J, Karayiorgou M et al: Magnitude and distribution of linkage disequilibrium in population isolates and implications for genome-wide association studies. Nat Genet 2006; 38: 556–560.
Abdellaoui A, Hottenga JJ, de Knijff P et al: Population structure, migration, and diversifying selection in the Netherlands. Eur J Hum Genet 2013; 21: 1277–1285.
O'Dushlaine CT, Morris D, Moskvina V et al: Population structure and genome-wide patterns of variation in Ireland and Britain. Eur J Hum Genet 2010; 18: 1248–1254.
Pardo LM, Piras G, Asproni R et al: Dissecting the genetic make-up of North-East Sardinia using a large set of haploid and autosomal markers. Eur J Hum Genet 2012; 20: 956–964.
Jakkula E, Rehnstrom K, Varilo T et al: The genome-wide patterns of variation expose significant substructure in a founder population. Am J Hum Genet 2008; 83: 787–794.
Green RC, Green JS, Buehler SK et al: Very high incidence of familial colorectal cancer in Newfoundland: a comparison with Ontario and 13 other population-based studies. Fam Cancer 2007; 6: 53–62.
Wang PP, Dicks E, Gong X et al: Validity of random-digit-dialing in recruiting controls in a case-control study. Am J Health Behav 2009; 33: 513–520.
Cronin S, Berger S, Ding J et al: A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum Mol Genet 2008; 17: 768–774.
Wellcome Trust Case Control Consortium, Craddock N, Hurles ME et al: Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 2010; 464: 713–720.
Li JZ, Absher DM, Tang H et al: Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008; 319: 1100–1104.
International HapMap 3 Consortium, Altshuler DM, Gibbs RA et al: Integrating common and rare genetic variation in diverse human populations. Nature 2010; 467: 52–58.
International HapMap Consortium: The International HapMap Project. Nature 2003; 426: 789–796.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Wickham H : ggplot2: Elegant Graphics for Data Analysis. Springer: New York, 2009.
Lawson DJ, Hellenthal G, Myers S, Falush D : Inference of population structure using dense haplotype data. PLoS Genet 2012; 8: e1002453.
Alexander DH, Novembre J, Lange K : Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009; 19: 1655–1664.
Patterson N, Moorjani P, Luo Y et al: Ancient admixture in human history. Genetics 2012; 192: 1065–1093.
Barrett JC, Fry B, Maller J, Daly MJ : Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics (Oxford, England) 2005; 21: 263–265.
Loh PR, Lipson M, Patterson N et al: Inferring admixture histories of human populations using linkage disequilibrium. Genetics 2013; 193: 1233–1254.
Raghavan M, Steinrucken M, Harris K et al: POPULATION GENETICS. Genomic evidence for the Pleistocene and recent population history of Native Americans. Science 2015; 349: aab3884.
Martin LJ, Crawford MH, Koertvelyessy T, Keeping D, Collins M, Huntsman R : The population structure of ten Newfoundland outports. Hum Biol 2000; 72: 997–1016.
Martijn CA : Early Mikmaq presence in Southern Newfoundland: an ethnohistorial perspective, c.1500-1763. Newfoundland and Labrador Studies 2003; 19: 44–102.
Historical Statistics of Newfoundland and Labrador Vol. I. Creative Printers & Publishers Limited, Office of the Queen's Printer: St John's, Newfoundland, 1970.
Young TL, Woods MO, Parfrey PS, Green JS, Hefferton D, Davidson WS : A founder effect in the Newfoundland population reduces the Bardet-Biedl syndrome I (BBS1) interval to 1cM. Am J Hum Genet 1999; 65: 1680–1687.
Aksentijevich I, Masters SL, Ferguson PJ et al: An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 2009; 360: 2426–2437.
Wright AF, Carothers AD, Pirastu M : Population choice in mapping genes for complex diseases. Nat Genet 1999; 23: 397–404.
Eaves IA, Merriman TR, Barber RA et al: The genetically isolated populations of Finland and Sardinia may not be a panacea for linkage disequilibrium mapping of common disease genes. Nat Genet 2000; 25: 320–323.
Taillon-Miller P, Bauer-Sardina I, Saccone NL et al: Juxtaposed regions of extensive and minimal linkage disequilibrium in human Xq25 and Xq28. Nat Genet 2000; 25: 324–328.
Acknowledgements
The study was supported by Newfoundland and Labrador Provincial RDC; Memorial University of Newfoundland; Canadian Institutes of Health Research. We acknowledge the International HapMap Consortium for the HapMap3 data and acknowledge all the donor communities and research groups of the Consortium listed in reference 24. We also acknowledge the genotyping of the NL samples conducted by Dr Stephen B Gruber, USC Norris Comprehensive Cancer Center, University of Southern California as part of the ColoRectal Transdisciplinary Study (CORECT). The study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from http://www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under award 076113 and 085475. The study also makes use of data generated from the study ‘A genome-wide association study of sporadic ALS in a homogenous Irish population’, which was funded by the Muscular Dystrophy Association (United States) (SC, DGB, OH), by the Irish Institute of Clinical Neuroscience Travel Award (SC), by the ALS Association and the Packard Center for ALS Research at Johns Hopkins and by the intramural programs of the National Institute on Aging (SB, JHD, JC, NW, DGH) and the National Institute on Neurological Disorders and Stroke (BJT), and the genotyping of samples was provided by the National Institute of Neurological Disorders and Stroke (NINDS). The data set(s) used for the analyses described in the manuscript were obtained from the NINDS Database found at https://dbgap.ncbi.nlm.nih.gov/aa/wga.cgi?page=request_details_pi&filter=arid&arid=70677 through dbGaP accession number 15035-2. The study also makes use of partial data generated from the Human Genome Diversity Project. The data set can be found at http://www.hagsc.org/hgdp/files.html.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Zhai, G., Zhou, J., Woods, M. et al. Genetic structure of the Newfoundland and Labrador population: founder effects modulate variability. Eur J Hum Genet 24, 1063–1070 (2016). https://doi.org/10.1038/ejhg.2015.256
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.256
This article is cited by
-
The Newfoundland and Labrador mosaic founder population descends from an Irish and British diaspora from 300 years ago
Communications Biology (2023)
-
Macrophage migration inhibitory factor may play a protective role in osteoarthritis
Arthritis Research & Therapy (2021)
-
The genetic architecture of Stargardt macular dystrophy (STGD1): a longitudinal 40-year study in a genetic isolate
European Journal of Human Genetics (2020)
-
Genetic associations in community context: a mixed model approach identifies a functional variant in the RBP4 gene associated with HDL-C dyslipidemia
BMC Medical Genetics (2018)
-
A common variant in CLDN14 causes precipitous, prelingual sensorineural hearing loss in multiple families due to founder effect
Human Genetics (2017)
