
Abstract
Variants in the WD repeat 45 (WDR45) gene in human Xp11.23 have recently been identified in patients suffering from neurodegeneration with brain iron accumulation, a genetically and phenotypically heterogeneous condition. WDR45 variants cause a childhood-onset encephalopathy accompanied by neurodegeneration in adulthood and iron accumulation in the basal ganglia. They have been almost exclusively found in females, and male lethality was suggested. Here we describe a male patient suffering from a severe and early neurological phenotype, initially presenting early-onset epileptic spasms in clusters associated with an abnormal interictal electroencephalography showing slow background activity, large amplitude asynchronous spikes and abnormal neurological development. This patient is a carrier of a 19.9-kb microdeletion in Xp11.23 containing three genes, including WDR45. These findings reveal that males with WDR45 deletions are viable, and can present with early-onset epileptic encephalopathy without brain iron accumulation.
Similar content being viewed by others


Introduction
Variants in the WD repeat 45 (WDR45) gene have recently been identified in patients suffering from neurodegeneration with brain iron accumulation (NBIA). NBIA is a genetically and phenotypically heterogeneous condition.1 Among all NBIA forms, beta propeller protein-associated neurodegeneration (BPAN) syndrome affects a subgroup of patient characterized by childhood-onset encephalopathy accompanied by neurodegeneration in adulthood and iron accumulation in the basal ganglia, notably in the globus pallidus or the substantia nigra.2 WDR45 variants associated with BPAN syndrome have been almost exclusively found in females and only three male cases have been described to date.3 This low incidence of male cases has been attributed to male lethality and the existence of a few male carriers to somatic mosaicism.2 Among published cases, 70% of patients suffered from epileptic seizures beginning after 3 months of age.3, 4
Early-onset epileptic encephalopathies (EOEE) form a group of severe epilepsies occurring in the first 3 months of life. Clinical signs include stormy seizures associated with an altered interictal electroencephalographic (EEG) pattern and abnormal neurological development. They can be classified according to interictal/ictal EEG pattern, type of seizures and age at onset. These epileptic disorders are highly heterogeneous in terms of aetiology as they can be symptomatic (eg, cortical malformation or stroke), or have a metabolic or genetic origin. Variants in several genes were shown to cause EOEE.5
During the genetic characterization of a large cohort of patients suffering from various forms of EOEE, we identified a de novo deletion of the WDR45 gene in a male patient. This patient suffers from a more severe and earlier form of BPAN syndrome. Our unexpected findings reveal that alterations of the WDR45 gene region in Xp11 are viable in male patients and can initially present as EOEE.
Subjects and methods
Subjects
A cohort of 352 EOEE patients was studied. The inclusion criteria were onset of epilepsy within the first 3 months of life, absence of any metabolic or obvious structural cause, abnormal interictal EEG and severe seizures that resisted to antiepileptic drugs. All patients with a normal karyotype were screened, using Sanger sequencing, for the presence of a variant in the most frequently mutated EOEE genes (ARX, KCNQ2, STXBP1, SCN2A, SCN8A) and, using array comparative genomic hybridization (array-CGH), to detect submicroscopic deletions/insertions. This project was approved by the local ethics committee (CPP Sud Méditerranée I, PHRC 2011/27).
Array-CGH and quantitative PCR
Array-CGH was performed using genomic DNA isolated from blood lymphocytes by standard methods. Array-CGH was performed using NimbleGen 3 × 720 K Whole Genome arrays (Roche NimbleGen, Madison, WI), providing an average resolution of 2.5 kb. Microarrays were hybridized, scanned and analysed according to the manufacturer’s instructions using a Roche Nimblegen microarray analysis platform. Regions showing abnormal dosage were compared with regions already known to represent non-pathogenic copy-number variations listed in the database of genomic variants (http://projects.tcag.ca/cgi-bin/variation/gbrowse/hg18). Quantitative PCR (qPCR) was performed using FastStart Universal SYBR Green Master (Rox; Roche). The Xp11.23 variant was submitted to ClinVar with accession number SCV000223701 (http://www.ncbi.nlm.nih.gov/clinvar/).
Results
Clinical phenotype
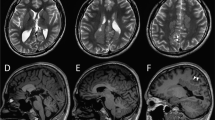
The patient was referred to our centre at the age of 3 months with epileptic spasms associated with focal seizures. He was born to healthy parents after normal delivery and he adapted well initially. Neurological examination, at 3 months of age, was very abnormal – no head control, no eye contact in a hypokinetic patient with global hypotonia. He had cortical blindness and ophthalmologic examination was normal (retina and anterior chamber). EEG showed high amplitude slow waves with multifocal spikes. A cluster of asymmetric spasms was recorded. At 7 months of age, brain MRI revealed a global loss of brain volume including supra and infratentorial areas, T2 hyperintensity of the posterior fossa (bilateral dentate nuclei) and lipid accumulation on spectroscopy (white matter, not shown). This slight peak of lipids at 1.3 p.p.m. was present within the white matter only at the initial MRI assessment. No iron accumulation could be detected at this age, though no gradient echo sequence and no susceptibility weighted images were performed (Figure 1). The evolution was marked by the absence of motor or sensory acquisitions. His neurological examination showed dystonic and spastic quadriplegia, a motionless child without spontaneous movement, bedridden with very poor eye contact. Head circumference remained normal throughout follow-up. At 19 months of age, brain MRI showed increased global brain atrophy with T2 hyperintensity of the white matter. Iron accumulation was only detected at 5 years of age, using a susceptibility weighted imaging (SWI) sequence, in striata, red nuclei and substantia nigra (Figure 1). Interestingly, conventional sequences (T1, T2) failed to detect iron accumulation at this stage (Figure 1). Blood (NH4, glycemia, amino-acid chromatography, lactates, pyruvates, biotinidase activity, pipecolic acid, transaminases, acid hydrolases), urine (chromatography of organic acids, muco/oligopolysaccarids, lactates) and cerebrospinal fluid (neurotransmitter profil, amino-acid chromatography, lactates, pyruvates) analyses were normal. Study of the mitochondrial respiratory complex on muscle was normal.

Brain MRI performed at 7 months, 19 months and 5 years of age. Axial T2 sequences at different ages failed to detect iron accumulation. SWI sequence (right image) reveals an hyposignal in the basal ganglia at the level of globus pallidus and substantia nigra (white arrows), bilaterally (red nuclei are not shown).
Molecular characterization of an Xp11.23 de novo microdeletion
High-resolution array-CGH analysis detected a 19.9-kb deletion in Xp11.23 hg18 chrX:g.(48 802 381_48 809 279)_(48 829 265_48 854 335)del (Figure 2a). Using parental DNAs, we found that this deletion occurred de novo. The deleted region contains three genes – WDR45, coiled-coil domain containing 120 (CCDC120) and prenylated rab acceptor 1 domain family member 2 (PRAF2; Figure 2b). The deletion was confirmed using qPCR and was not in a mosaic status in the patient (Figure 2c). Only one study reported a variation of Xp11.23 in the database of genomic variants (see Subjects and methods above) but no information is available for the patient gender.

Characterization of the deletion (a). Detection of a 19.9- kb deletion on the X chromosome using array-CGH. The grey box shows the position of the deleted region extending from 48 809 279 to 48 829 265 pb (NCBI36/Hg18). The experiment was performed using dye swap and the results of Cy5 labelling (upper tracks) and Cy3 labelling (lower tracks) are shown for the same patient. (b) Schematic representation of the Xp11.23 region showing the deletion identified in the BPAN patient. The grey bar shows the deleted region. Exons are represented as black, grey or white boxes for WDR45, CCDC120 and PRAF2 genes respectively. Arrows indicate the orientation of the transcription and the position of the translation initiation codon. (c) Quantification of WDR45 genomic DNA targets using qPCR. The control female DNA was arbitrarily set at 1 and each experiment was performed in triplicate. The mean of the three experiments is provided as a number above each condition.
Discussion
The patient described here is the first male case presenting a constitutional WDR45 deletion in the context of EOEE. Several brain MRIs failed to detect signs of iron accumulation using conventional sequences, even several years after the symptoms onset, because no SWI or gradient echo sequences were acquired until 5 years of age. Consecutively, SWI/GRE should be included when imaging a child with unexpected dystonia. Early images rather revealed global brain atrophy, a nonspecific finding in many cases of EOEE. This patient thus presented a peculiar form of BPAN beginning with severe epilepsy and cortical blindness. In BPAN, the vast majority of published cases are females, with only three reported male patients. It has been hypothesized that the gender bias was due to WDR45 variants lethality in males.3 Along the same line, somatic mosaicism would explain the similar phenotype between the few affected males and female patients. Indeed, Haack et al2 have confirmed the presence of somatic mosaicism in a male patient. However, the presence of a mosaic variant was not demonstrated but simply inferred for the two other males. The patient described in this report is not mosaic, at least in his lymphoblasts, and this could explain the early disease onset. We hypothesize that the initial clinical presentation of a WDR45 deletion is an EOEE-like syndrome, followed a few years later by a more severe BPAN syndrome. Interestingly, despite full WDR45 deletion, iron accumulation was only detected at 5 years of age. By comparison, the youngest female in whom iron accumulation was first detected was 11 years old6 suggesting that non-mosaic males have a much earlier phenotype of iron deposition.
We cannot exclude the involvement of the two additional deleted genes and this peculiar clinical presentation could be due to a contiguous gene syndrome. In fact, the PRAF2 gene encodes a member of the PRA1 domain-containing protein family. This family is already involved in the regulation of glutamate transport7, 8 whose defects are implicated in epilepsy.9 The function of PRAF2 is unknown, and the corresponding protein has a brain expression and is enriched in synaptic vesicles.10 CCDC120 mediates neurites' outgrowth via the regulation of cytohesin-2.11 Hence, CCDC120 deletion could also be an additional mechanism as abnormal formation of the neuronal network may contribute to intellectual deficiency or the abnormal motor and sensory acquisitions in this patient. The role of PRAF2 and CCDC120 during brain development and in genetic diseases remains to be investigated. Nonetheless, whatever their function is, our work reveals that males with deletions of WDR45 are viable, and can present with EOEE.
References
Gregory A, Hayflick SJ : Genetics of neurodegeneration with brain iron accumulation. Curr Neurol Neurosci Rep 2011; 11: 254–261.
Haack TB, Hogarth P, Kruer MC et al: Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet 2012; 91: 1144–1149.
Hayflick SJ, Kruer MC, Gregory A et al: β-Propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain 2013; 136: 1708–1717.
Saitsu H, Nishimura T, Muramatsu K et al: De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet 2013; 45: 445–449.
Lin CI, Orlov I, Ruggiero AM et al: Modulation of the neuronal glutamate transporter EAAC1 by the interacting protein GTRAP3-18. Nature 2001; 410: 84–88.
Ohba C, Nabatame S, Iijima Y et al: De novo WDR45 mutation in a patient showing clinically Rett syndrome with childhood iron deposition in brain. J Hum Genet 2014; 59: 292–295.
Mastrangelo M, Leuzzi V : Genes of early-onset epileptic encephalopathies: from genotype to phenotype. Pediatr Neurol 2012; 46: 24–31.
Liu Y, Vidensky S, Ruggiero AM, Maier S, Sitte HH, Rothstein JD : Reticulon RTN2B regulates trafficking and function of neuronal glutamate transporter EAAC1. J Biol Chem 2008; 283: 6561–6571.
Meldrum BS, Akbar MT, Chapman AG : Glutamate receptors and transporters in genetic and acquired models of epilepsy. Epilepsy Res 1999; 36: 189–204.
Koomoa D-LT, Go RCV, Wester K, Bachmann AS : Expression profile of PRAF2 in the human brain and enrichment in synaptic vesicles. Neurosci Lett 2008; 436: 171–176.
Torii T, Miyamoto Y, Tago K et al: Arf6 guanine nucleotide exchange factor cytohesin-2 binds to CCDC120 and is transported along neurites to mediate neurite growth. J Biol Chem 2014; 289: 33887–33903.
Acknowledgements
We thank the patient and his family for their participation in this study. This work was supported by Programme Hospitalier de Recherche Clinique 2011, Inserm and Aix Marseille University. We thank the Centre de Ressources Biologiques of La Timone Children's Hospital (Karine Bertaux, Cécile Mouradian and Andrée Robaglia-Schlupp) for assistance with the samples used in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Abidi, A., Mignon-Ravix, C., Cacciagli, P. et al. Early-onset epileptic encephalopathy as the initial clinical presentation of WDR45 deletion in a male patient. Eur J Hum Genet 24, 615–618 (2016). https://doi.org/10.1038/ejhg.2015.159
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.159
This article is cited by
-
A comprehensive phenotypic characterization of a whole-body Wdr45 knock-out mouse
Mammalian Genome (2021)
-
Autophagy and disease: unanswered questions
Cell Death & Differentiation (2020)
-
A Novel and Mosaic WDR45 Nonsense Variant Causes Beta-Propeller Protein-Associated Neurodegeneration Identified Through Whole Exome Sequencing and X chromosome Heterozygosity Analysis
NeuroMolecular Medicine (2019)
-
WDR45 mutations in three male patients with West syndrome
Journal of Human Genetics (2016)
