
Abstract
Molecular genetic testing for hereditary hemochromatosis (HH) is recognized as a reference test to confirm the diagnosis of suspected HH or to predict its risk. The vast majority (typically >90%) of patients with clinically characterized HH are homozygous for the p.C282Y variant in the HFE gene, referred to as HFE-related HH. Since 1996, HFE genotyping was implemented in diagnostic algorithms for suspected HH, allowing its early diagnosis and prevention. However, the penetrance of disease in p.C282Y homozygotes is incomplete. Hence, homozygosity for p.C282Y is not sufficient to diagnose HH. Neither is p.C282Y homozygosity required for diagnosis as other rare forms of HH exist, generally referred to as non-HFE-related HH. These pose significant challenges when defining criteria for referral, testing protocols, interpretation of test results and reporting practices. We present best practice guidelines for the molecular genetic diagnosis of HH where recommendations are classified, as far as possible, according to the level and strength of evidence. For clarification, the guidelines’ recommendations are preceded by a detailed description of the methodology and results obtained with a series of actions taken in order to achieve a wide expert consensus, namely: (i) a survey on the current practices followed by laboratories offering molecular diagnosis of HH; (ii) a systematic literature search focused on some identified controversial topics; (iii) an expert Best Practice Workshop convened to achieve consensus on the practical recommendations included in the guidelines.
Similar content being viewed by others

Aim and scope
The aim of the current guideline is to provide recommendations on: (i) criteria for testing, (ii) strategies for testing and (iii) reporting results for the molecular genetic testing of hereditary hemochromatosis (HH). It is intended to guide decisions related to genetic testing and interpretation of test results for clinical biochemists, geneticists, genetic counselors, physicians, other health-care providers, individuals with suspected or confirmed HH and their family members.
Bakground
Classification and pathophysiology
HH is a term generally used to define a group of autosomal recessive genetic disorders characterized by iron accumulation in parenchymal organs, primarily the liver, which can potentially result in impaired organ structure and function. In recent years, hepcidin has been identified as the key hormone that controls the saturation of plasma transferrin with iron (transferrin saturation). Hepcidin deficiency causes increased transferrin saturation, which is the unifying feature and principal biochemical finding of all forms of HH. Homozygous or compound heterozygous defects in the hepcidin gene (HAMP) and in genes that stimulate hepcidin production such as HFE, HJV and TFR2 are therefore associated with hemochromatosis. Ferroportin disease is not classified as hemochromatosis because it is classically not associated with high transferrin saturation (TS) or low hepcidin concentrations (reviewed in Fleming and Ponka1). It is an autosomal dominant disorder caused by genetic defects in SLC40A1, which in its classical form, is associated with loss of its exporting function and consequent hepatic and splenic iron overload. In some rare cases, however, iron overload may occur in patients carrying SLC40A1 gene defects with a gain of its exporting function. In this case of non-classical ferroportin disease, patients may exhibit a hemochromatosis phenotype and are classified as HH.2 A summary of HH classification, nomenclature, pathogenesis and major clinical features is provided in Table 1.
HFE-related HH
By far the most common, well-defined and prevalent form of HH is HFE-related HH, associated with homozygosity for the A allele in the single nucleotide variant rs1800562, NG_008720.2:g.10633G>A; NM_000410.3:c.845G>A; NP_000401.1:p.Cys282Tyr), most commonly referred in the literature as C282Y (p.C282Y according to the recommended nomenclature). This form of HH is particularly common in Caucasians where 1 in 200–300 individuals are homozygous for p.C282Y (reviewed in European Association for the Study of Liver,3 Bacon et al4 and van Bokhoven et al5). The disease’s penetrance is low, affecting predominantly males between 40 and 60 years of age. Despite the remarkable genetic homogeneity of HFE-related HH, the clinical expression is variable, and 75–85% of p.C282Y homozygous individuals do not develop the disease.2 This low penetrance of the genotype emphasizes the need to better define genetic modifiers and environmental factors which contribute to iron overload and to severe clinical expression in these individuals.
Epidemiology, clinical diagnosis, management and treatment of HH was recently reviewed and discussed in several published practice guidelines and reports.3, 4, 5, 6, 7, 8, 9 Those guidelines were specifically directed to the common HFE-related HH and do not include the rarer non-HFE-related forms of HH for which there are still sparse clinical and epidemiological data available, so that their biochemical or clinical penetrance is still not known.
Rare forms of HH and ferroportin disease
The rare forms of HH (reviewed in Mayr et al,2 Camaschella and Poggiali,10and Brissot et al11), generally classified as Rare Hereditary Hemochromatosis (http://www.orpha.net/consor/cgi-bin/Disease_Search_Simple.php) comprise the non-HFE-related HH forms such as juvenile hemochromatosis, associated with variants in the hemojuvelin (HFE2 or HJV) or hepcidin (HAMP) genes, and hemochromatosis of adult onset resulting from variants in the gene for transferrin receptor 2 (TFR2). In addition to these ‘non-HFE-associated hemochromatosis’ variants, rare HFE variants in combination with p.C282Y have been reported12 as well as the homozygous HFE deletion which is a common cause of HH in Sardinia.13 As discussed above, genetic defects in ferroportin (SLC40A1) are associated with ferroportin disease, an autosomal dominant disorder of iron overload not generally classified as HH except when associated with gain of function variants.
Some rare forms of HH, as well as ferroportin disease, are increasingly found in patients who have been clinically characterized as HH and that test negative for p.C282Y homozygosity in HFE. They present with phenotypically proven hepatic iron overload with no other explanation. They have a much broader geographical distribution than the classical HFE-related HH, namely in Asian populations where they may represent the main non-hematological cause of hereditary iron overload.14, 15, 16 This fact has important implications for practical molecular diagnostic strategies in countries where the allele frequency of the p.C282Y variant of the HFE gene is low.
In addition to the genes described above, further genes have been associated with iron overload. A novel variant in the Iron-Responsive Element motif of the H-ferritin mRNA was described in a Japanese family affected by dominantly inherited iron overload,17 but the impact of this variant as a more widespread cause of HH in other populations is still not known. Furthermore, genes encoding endogenous regulators of hepcidin expression, such as the bone morphogenetic protein 6 (BMP6) or the SMAD 6 and 7 proteins are candidates for non-HFE-related hemochromatosis in humans.18, 19 So far, however, there are no described cases of HH associated with variants in any of these proteins.
Other genetic iron overload disorders unrelated to HH
It is important to note that other genetic causes of severe iron overload exist, but these should be distinguished from HH because of their distinct clinical and pathological presentation. These include iron-loading anemias due to ineffective erythropoiesis (thalassemia syndromes, hemoglobinopathies, sideroblastic anemias, congenital dyserythropoietic anemia) or due to hemolysis, defects in iron acquisition by the erythroid precursors (hypotransferrinemia, defect in DMT1, aceruloplasminemia (reviewed in Fleming and Ponka1and Donker et al20) or repeated blood transfusions. All these disorders demand a distinct clinical and molecular diagnosis workup that is out of the scope of the present document.
Clinical presentation and reasons for genetic testing referral
Early symptoms of HH are non-specific and may include weakness, lethargy, weight loss and arthralgia. Other suggestive, but also not specific, signs of more advanced disease include skin pigmentation, liver cirrhosis, chondrocalcinosis, arthritis, diabetes, hypogonadism, hepatocarcinoma or cardiomyopathy. If any of these conditions is related to HH, they will be necessarily associated with biochemical evidence of iron overload, namely an increased TS and elevated serum ferritin. Biochemical disease expression is usually present in presymptomatic disease stages, where elevated TS is the first indicator of iron overload. Therefore, patients with consistently increased TS and serum ferritin (SF) in the absence of hematological diseases are suspected to have HH and should be referred for molecular genetic testing. HFE genotyping is the first step for diagnosis, confirming or excluding the diagnosis of HFE-related HH. In patients presenting with phenotypically proven iron overload (ie, with severely increased liver iron stores demonstrated by biopsy, magnetic resonance imaging or quantitative phlebotomies) and a negative test for p.C282Y homozygosity, genetic testing for other HH-related variants is indicated if other hepatic or hematological causes have been excluded. This should be requested from referral centers offering expert advice on iron overload disorders and appropriate molecular diagnosis.
Particular types of non-HFE-related HH have specific phenotypic signs that allow a rational approach to genetic testing in patients with suspected non-HFE HH. Juvenile hemochromatosis (Hemochromatosis type 2) is generally characterized by its early onset and a particularly severe phenotype.21 Patients with juvenile hemochromatosis typically present before the age of 30 with severe systemic iron overload, heart failure and hypopituitarism as common clinical manifestations. TFR2-related HH is clinically similar to HFE-related HH and may be suspected in any phenotypically proven HH adult patient, and even younger patients,22 in the absence of HFE p.C282Y homozygosity.23 A well-documented unexplained classical iron overload phenotype in a patient found heterozygous for p.C282Y should raise the suspicion of other rare variants/deletions in HFE.24 The classical ferroportin disease (type A) is characterized by a dominant mode of transmission and hepatic and spleen iron overload mainly in Kupffer cells and macrophages (because of ‘loss of function’ variants) reflected by hyperferritinemia with high/normal or low TS.25 This disease requires a more careful differential diagnosis with other very common causes of hyperferritinemia such as the Dysmetabolic Iron Overload Syndrome associated with insulin resistance and liver steatosis.26 The non-classical form of ferroportin disease (type B), also with an autosomal dominant mode of transmission, is characterized by additional hepatocellular iron deposits and high TS (due to ‘gain of function’ variants) and requires a differential diagnosis with other forms of HH.27
Strategies for enhanced case detection
Because of the unspecific nature of the reported signs or symptoms, effective case detection in HH is largely dependent on a strong clinical awareness and motivation to request the appropriate biochemical tests and subsequent genetic testing in suspected cases. The initial approach to diagnosis in any suspected case is the determination of TS. Although not absolutely necessary, it is advisable to confirm elevated TS with a second determination on a fasting specimen.4 It should be noted that, in spite of being the most widely accepted screening test for systemic iron overload and a very well-standardized laboratory parameter, there is no universally defined TS cutoff value to identify patients eligible for further testing and a variety of cutoff values have been used in different studies (reviewed in European Association for the Study of Liver28). A review of studies in several populations using cutoffs of TS ranging from 45 to 60% was presented in Table 5 of the 2010 EASL Clinical Practice Guidelines for HFE Hemochromatosis28 and the positive predictive value of elevated TS for the detection of p.C282Y homozygotes was estimated to vary from 4.3 to 21.7%.28 Besides TS, SF is a surrogate of iron stores and hyperferritinemia is an indicator of iron overload, when inflammatory/malignant conditions are excluded. Although SF is a well-standardized laboratory parameter, there is also no universally accepted reference range. A review of studies in several populations using cutoffs of SF ranging from 250 to 428 μg/l in males and from 130 to 302 μg/l in females is also shown in Table 5 of the 2010 EASL Clinical Practice Guidelines for HFE Hemochromatosis, showing a positive predictive value for detecting p.C282Y homozygosity ranging from 1.6 to 17.6%.28 It should be stressed that SF can be influenced by local environmental factors such as the lifestyle habits of the population (particularly alcohol consumption, weight gain and sedentary lifestyle) or geographical region.29 Ideally, cutoff values should be always established according to local values for the upper limit of the reference range.29 If SF is used as a predictor of HH, it should also be noted that it can be elevated in the absence of increased iron stores in patients with chronic inflammatory conditions, particularly in patients with liver disease (alcoholic liver disease, chronic viral hepatitis and non-alcoholic fatty liver disease).4 Studies have shown that patients with hyperferritinemia are less likely to be p.C282Y homozygotes if they have abnormal liver transaminase activities,30 this paradox being explained by the low yields of hemochromatosis screening reported by some liver clinics. In this context, it should be noted that iron overload is not the most common cause of an elevated ferritin, and the finding of hyperferritinemia without an increased TS is not a criterion for HFE genetic testing, demanding a different approach for differential diagnosis.31
When a diagnosis of HFE-related hemochromatosis is established in a proband, genetic screening of asymptomatic siblings, or other first-degree relatives, is a cost-effective strategy to enhance case detection and early diagnosis.32, 33 Genetic screening of the general population is not recommended because of the low penetrance of the disease. Although biochemical evidence of iron overload usually precedes symptomatic disease, biochemical population screening for iron overload is also not recommended because large studies have failed to show its cost-effectiveness.34, 35 However, the true prevalence of symptomatic HH in the general population, in particular in European populations, is also unknown, which increases the uncertainty of cost-efficiency analyses.
Methodology and results
To achieve a wide expert consensus for these guidelines, a drafting group compiled the evidence to support recommendations, prior to a wider consultation. In that context, a survey on the current practices used by laboratories offering molecular diagnosis of HH was undertaken, as well as a systematic literature search focused on some identified controversial topics. Finally, a group of clinicians and scientists involved in HH diagnostics and iron disorders, supported by the European Molecular Genetics Quality Network (EMQN), met on 14–15 May 2014 at a workshop in Porto, Portugal to consider the collected evidence and to formulate best practice guidelines. Discussions at the workshop focused on testing criteria and protocols, interpretation of results and reporting. Consensus guidelines were established. To comply with current standards for evidence assessment, recommendations have been weighted according to the Grading of Recommendations Assessment, Development and Evaluation system (GRADE).3, 36 If the level of published evidence was very low, decisions were taken based on the experts’ opinion.
Results of the survey on current practices of genetic diagnosis of HH
In order to assess current practices on the molecular diagnosis of HH, a survey was conducted by means of a standard questionnaire posted online (using the SurveyMonkey platform) by the European Molecular Quality Network (EMQN) from the 20 April to 8 June 2012 and advertised not only among EMQN members, but also through a network of clinical and research experts on hemochromatosis and other iron overload disorders, promoted by the European Federation of the Associations of Patients with Hemochromatosis (EFAPH). For the recruitment process, a link to the online survey was emailed out to all the groups mentioned above asking them to participate. For the purpose of analysis, a pre-analytical validation was performed with exclusion of incomplete questionnaires or respondents who did not declare to perform testing for HFE variants. Descriptive statistical methods were used to report quantitative data, and content analysis was used for the replies to the open questions. Validated questionnaires were obtained from a total of 113 participating laboratories from 26 different countries (Australia, Austria, Belgium, Canada, Cyprus, Czech Republic, Estonia, France, Germany, Greece, Hungary, India, Ireland, Israel, Italy, Latvia, Netherlands, Norway, Poland, Portugal, Slovakia, South Africa, Spain, Sweden, Switzerland and United Kingdom). The vast majority of responding laboratories were Clinical Diagnostic Laboratories (89%), 75% of these were members of the EMQN and 10% participated in other Quality Control schemes either International Quality Networks (UKNEQAS, Equalis) or local External Quality Control. A small proportion of respondents (4%) represented research laboratories and 7% self-reported as Reference Centers. Participants routinely perform HFE genotyping either at a local (36%), and/or regional (49%), and/or national (44%) and/or international (13%) levels. The average volume of requests in the majority of laboratories (52%) is more than 200 per year, in half of these being reported a volume of more than 500 requests per year. In addition to standard p.C282Y/p.H63D HFE genotyping, 22% (n=25) of responding laboratories are also offering full gene sequencing for HFE and the other iron genes required for the diagnosis of non-HFE-related HH. Most of the laboratories offering these tests receive requests at a national (72%) or international (28%) level. Some of the laboratories (n=10) reported to test for non-HFE HH an average of more than 20 tests/year, two of them receiving more than 100 or 200 requests/year (respectively, in UK and France). The remaining reported a volume of 6–20 (n=10) or 1–5 (n=5) tests/year.
Current practices on ‘criteria for testing’
The majority of participants in the survey reported that HFE genotyping is mainly requested for diagnosis of a suspected index case (ranging from 50 to 75% of requests) and the number of requests for predictive (asymptomatic) or carrier testing varies from less than 10 to 50% of all requested tests. The majority of the participating laboratories do not have specific prerequisites for HFE genotyping indicated in the referral form, accepting for testing all requests with a valid clinical referral. Some laboratories (27%) provide in their referral forms specific testing prerequisites which are established either at local (43%), regional (23%) or national (43%) levels. Regarding the practice of testing for non-HFE-related HH, the majority (64%) of the laboratories offering these tests reported to have defined strategies for the selection and order of tests, but only a few (n=6) refer to published guidelines available on their national reference websites. Prerequisites indicated in the referral form are used in some laboratories (n=9) which were either established locally (n=3) or at a national level (n=6).
In general, referrals for testing that do not meet the established criteria are accepted or not according to different local policies. When asked about their practice regarding the follow-up of missing clinical prerequisites, the majority of respondents report contacts with the referring clinician either by telephone or E-mail (45%) or sending back the referral form asking for the requisites (36%). The remaining respondents declared that they searched for information on other available databases (9%), simply accepted the requests (9%) or added comments on the report form (18%).
When asked about the number of HH cases identified with p.C282Y homozygosity, one-third of respondents (33%) reported a detection rate of less than 5% and only a minority (13%) reported a detection rate superior to 20%. The majority (54%) reported a detection rate between 5 and 20%. This rate is similar to the reported prevalence of p.C282Y homozygosity among patients presenting with elevated TS in several primary health-care surveys.28 No significant differences were found in terms of detection rates between laboratories who established prerequisites in the referral forms and those who do not have written prerequisites, suggesting that the practice of using prerequisites does not necessarily increase efficiency. Owing to its rarity and complexity, efficacy regarding the diagnosis of non-HFE-related HH was not addressed in this survey.
Current practice on ‘methods and testing strategies’
Methods used for HFE testing
All the laboratories responding to this survey routinely screen for the HFE variants p.C282Y and p.H63D, although some of them (12%) report to have a policy of testing for p.H63D only in the situation of clinically affected patients with a previous result showing p.C282Y heterozygosity. In addition, 64% of respondents also test routinely for the p.S65C variant, some of them reporting that the test is performed occasionally and only on specific demands. A minority of laboratories (6%) reported to also routinely test for other genetic variants included in commercially sourced kits.
A large number of different methods are used by laboratories. The four methodological approaches most frequently reported were: real-time PCR (41%), PCR and RFLP (16%), PCR and reverse hybridization (14%), and direct sequencing (11%). In addition, other less commonly used (4.5–5.5%) include: allele-specific PCR, PCR and high-resolution melting or single-strand conformational polymorphism, pyrosequencing and single base extension. According to this survey, allelic discrimination real-time PCR was the most commonly described method for HFE genotyping.
About the need to test for p.H63D for the diagnosis of HFE-related HH
When questioned about the ‘real need’ and implications of testing for p.H63D, no consensual opinions were obtained among respondents. Some respondents declared serious doubts as to whether they should or should not test for p.H63D (16.7%), whereas others declared unambiguously that there is no need to test for p.H63D (19.4%), the most common arguments being that it is not clinically relevant, it has a high frequency in the normal population with uncertain implications, or that results may raise anxiety unnecessarily. The fact that they go on offering this test is, in many cases, explained by the pressure of requesting clinicians. The majority of laboratories (48.6%), however, still considered that testing for p.H63D is needed and/or important, although some of them stressed the importance of a careful wording on reports, which should be put in context, particularly in the absence of p.C282Y. The most consistent justification for p.H63D testing was the increased risk of iron overload in p.C282Y/p.H63D compound heterozygotes, a minority also admitting a risk for p.H63D homozygotes. Some pointed to the importance of giving lifestyle recommendations in these cases.
Methods used for non-HFE-related HH testing
A total of 25 laboratories reported to perform full gene sequencing for the diagnosis of non-HFE-related HH, the majority of these (52%) offering a panel of tests for HJV, HAMP, TFR2, SLC40A1 (FPN) and HFE-whole gene. Some laboratories (24%) also offer testing for FTH1 (for hyperferritinemia), and two of the responding laboratories perform a more complete panel of tests for other iron overload-related genes adding 5’UTRFTL (for hyperferritinemia), TF (for hypotransferrinemia), SLC11A2 (for anemia and iron overload), CP (for aceruloplasminemia) and BMP6 (so far not associated to human HH). The most common method used is direct DNA sequencing of the coding regions of selected genes, some laboratories reporting other methods such as single-strand conformation polymorphism analysis followed by direct sequencing of the detected variants and next-generation sequencing (NGS)-based approaches. Multiplex ligation-dependent probe amplification has been used in cases where a large gene deletion is suspected.
Current practices on ‘reporting’
The reporting of HFE genotyping results is generally performed by standard pre-defined templates, most commonly (63%) adapted according to specific reasons for referral. The reporting models and interpretative comments included are usually based on established guidelines, some laboratories referring to more general guidelines for genetic testing37, 38, 39, 40, 41 but the majority referring to specific HH guidelines established either at a national level42, 43, 44, 45, 46 or from other published clinical4, 6, 7, 8, 9, 28 or molecular diagnosis guidelines.47 The majority of respondents (69%) recommend or suggest predictive/carrier testing for first-degree relatives of detected cases, the majority of these (88%) specifically recommending genetic counselling for that purpose. Among respondents who recommend predictive/carrier testing to relatives of patients with HFE variants, 16% recommend it only for first-degree relatives of p.C282Y homozygous patients and 57% also recommend for relatives of p.C282Y heterozygous carriers. In addition, 28% of the respondents also recommended predictive/carrier testing to relatives of patients carrying any allele variant combination, including p.H63D and sometimes p.S65C. About one-third of respondents (31%) do not recommend in their reports predictive/carrier testing of relatives of patients with identified HFE variants.
Current practices on testing minors and value of risk assessment by testing spouses
When questioned about their policy regarding testing of minors for HFE-related HH, the majority of respondents (81%) declared to have a general policy of not testing minors (recommending postponement of the genetic test until the subject has reached the age of legal consent) or testing ‘young’ subjects (not younger than 16 years) as long as they understand the meaning and implications of the test. Exceptions to the rule were rare cases referred for diagnosis when there is a clear clinical indication, in conditions of altered iron status or evidence of liver disease, and when requested by a specialist (clinical hematologist or clinical geneticist). The remaining 19% of respondents declared to test samples from minors whenever requested, deferring responsibility to the requesting physician and written consent from the parents. When questioned about the value of offering testing to spouses to assess risks in offspring, opinions were divided. Opinions ranged from the total rejection, considering it useless or nonsense, to the acceptance as an important testing strategy. Arguments used in favor were mainly as a question of cost-effectiveness, because ‘testing one spouse is cheaper than testing all the children’ and the possibility of reassurance to offspring if the spouse does not carry p.C282Y, avoiding further follow-up arrangements. Some respondents also referred to satisfying couples who actively seek genetic advice and ask for that test, and others to the importance of having extended pedigree information for phenotype interpretation. Arguments against testing spouses included the inappropriate waste of resources because it is a late-onset disease and offspring can be offered advice at the appropriate time; the problems raised by false paternity findings; and variable penetrance of the p.C282Y/p.C282Y and p.C282Y/p.H63D genotypes, making it difficult to predict outcomes for offspring who therefore should be monitored.
Results of a systematic literature search on controversial topics
The expert team drafted a document that served as the discussion basis for the final guidelines. For most issues addressed, the quality of evidence and strength of recommendations had been already assessed elsewhere9, 28 according to the system developed by the Grading of Recommendations Assessment, Development and Evaluation (short GRADE) Working Group (http://www.gradeworkinggroup.org/index.htm). For consistency, those scores were used in this guidelines document accordingly. For some controversial topics, however, we performed a systemic review of the literature. These topics were: (i) The clinical value of testing for p.H63D; (ii) Role, efficacy and efficiency of cascade screening; and (iii) Follow-up of unaffected p.C282Y homozygotes and p.C282Y/p.H63D compound heterozygotes. For the systematic review, research questions were formulated for each topic. For each specific question, search terms were phrased as PICO (Patient, Intervention, Control and Outcome) that guided the searches in both MEDLINE and EMBASE using Medical Subject Headings (MESH) and free text words. Searches were limited to those written in English, but were not restricted to date.
The clinical value of testing for p.H63D
This was a pending question since the publication of the previous guidelines for the molecular genetic diagnosis of Type I (HFE-related) HH29 where the authors raised the question of an eventual call for an ‘iron overload’ genetic screen as opposed to an HFE genetic screen. As evidenced by the present survey results concerning the question about testing for p.H63D, there is still much confusion among clinicians and laboratory scientists regarding the use of HFE variants (particularly p.H63D) as disease susceptibility markers.
In order to determine the diagnostic utility of p.H63D testing, a systematic review of the literature was performed. Search terms according to the following PICO were phrased: Patients (P) were individuals with hemochromatosis or patients with iron overload. Intervention (I) was genotyping of the p.H63D variant of the HFE gene. Controls (C) were newborns or population based screening and Outcome (O) was the report of genotype frequency. With this search strategy, 931 references were retrieved. From these, 33 studies were selected as case–control studies where the hemochromatosis case definition was considered appropriate. In addition, 37 population-based screening studies were selected.
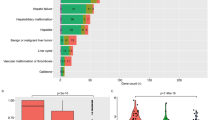
The association with hemochromatosis of the p.H63D variant in HFE (rs1799945, NG_008720.2:g.8671C>G; NM_000410.3:c.187C>G; NP_000401.1:p.His63Asp) was in question since its first description.48 Subsequent case–control studies have reported a median p.C282Y/p.H63D compound heterozygous genotype frequency among clinically characterized hemochromatosis cohorts of 4.6% (0–21.7%),48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78 which is significantly higher than the compound heterozygote frequency of 1.7% (0–7.7%) reported in screening studies.60, 63, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109 Although this observation suggests that compound heterozygosity is associated with hemochromatosis, longitudinal studies have shown that the risk of disease progression is low in compound heterozygotes identified by screening.110 A more recent report of the evolution of iron indices in 180 compound heterozygotes for p.C282Y and p.H63D identified in the HealthIron study111 concluded that in these, the mean iron indices do not change during middle age in males but SF increases in females. Although maintaining elevated iron indices during middle age, documented iron overload-related disease in compound heterozygotes is rare. Furthermore, p.C282Y/p.H63D compound heterozygote hemochromatosis patients with clinical disease expression frequently have additional risk factors for iron overload or liver disease.112, 113, 114, 115 In conclusion, although p.C282Y/p.H63D compound heterozygosity is a risk factor for slightly higher serum iron parameters and mildly increased hepatic iron stores,112, 116, 117, 118 this genotype is considered insufficient to cause hemochromatosis. Likewise, homozygosity for p.H63D is also rarely associated with hemochromatosis and also not considered to be a disease-associated genotype.119 This is the reason why genotyping for p.H63D is not reimbursed in France.
Role, efficacy and efficiency of cascade screening
The responses obtained in the survey to the question ‘to whom do you recommend predictive or carrier testing?’ disclosed a strong divergence in reported practices. To address this question, a systematic literature search was performed. Search terms were phrased as PICO where (P) was defined as patients with HFE-hemochromatosis; interventions (I) were screening by diagnostic test, measurement of serum iron parameters (TS, ferritin and/or testing for p.C282Y (p.H63D) variants in the HFE-gene; compared (C) with no screening; and the expected outcome (O) was cost, versus prevention of iron overload and prevention of symptoms. All systematic reviews or meta-analyses, comparative designs (cohort studies, case–control studies and cross-sectional studies) and observational studies were eligible for inclusion. A total of 180 publications were identified, of which only 14 were scored as being relevant to answer the research question.
Cascade or family screening in HH can be defined as a genotypic screening strategy that targets relatives of previously identified p.C282Y homozygotes. It is offered to individuals with an increased risk, usually with some knowledge about the disease, and is aimed to detect covert homozygotes. In general, it can be considered as less efficacious (proportion of covert homozygotes detected) but more efficient (proportion of covert homozygotes detected in relation to the screening effort) than population screening, and requires the availability of an index case.121
Data from controlled and uncontrolled family studies demonstrate that the majority of p.C282Y homozygotes detected by screening of first-degree relatives of homozygous patients have elevated ‘iron parameters’ more commonly in men than in women.33, 122, 123, 124, 125, 126, 127 Data from uncontrolled family studies indicate that ‘morbidity’ in p.C282Y homozygous family members of HH patients is considerable, with up to 42% of them having fibrosis or cirrhosis.33, 127, 128 This is confirmed by some, but not all, controlled studies that found an elevated risk of diseases (particularly rheumatic disorders) in family members of patients with HH.122, 129, 130
Data are not consistent on whether iron parameters of relatives of clinically detected p.C282Y homozygotes differ from relatives of p.C282Y homozygotes detected by genotyping,124, 126 whereas disease penetrance in p.C282Y homozygotes identified through family screening was not found to differ from those identified by population screening in both studies on this subject.123, 124 Finally, we retrieved two studies showing that mortality of parents, siblings and children of p.C282Y homozygotes identified by clinical presentation or other means is not higher than that in the general population.122, 131
From a study of 291 children of p.C282Y homozygotes, Adams132 recommended that the most cost-effective strategy is to screen spouses to avoid unnecessary investigation of offspring of probands. In another (hypothetical) cohort of siblings and children of an affected proband, and assuming that 80% of HH subjects accumulate iron (of whom 50% develop tissue damage), El-Serag32 compared the cost benefit analysis of no screening against four screening strategies that incorporate HFE testing or serum iron indices. They conclude that in family screening, it is more cost-effective: (i) to screen the spouse before children when testing two or more children, (ii) to screen the spouse when children are below the age of consent and (iii) to use HFE testing instead of serum iron indices. However, it should be noted that the conclusions of this study strongly depend, among other factors, on the model assumptions for the prevalence of p.C282Y carriers in the population, the risk for p.C282Y homozygotes to develop iron overload and complications and the setting (country).
The prevalence of p.C282Y homozygosity among siblings (~25%), offspring (~5%) and parents (~5%) of p.C282Y homozygous probands is increased in comparison with the general Caucasian population (~0.5%). If we estimate the average risk of the development of severe morbidity (eg, liver cirrhosis) for a p.C282Y homozygote as being 4% (5.6% in males and 1.9% in females),123 the morbidity risk for siblings and offspring of a p.C282Y homozygous proband can be calculated as 1.0% and 0.2%, respectively. This implies that 100 siblings, 500 children and 500 parents, respectively, need to be screened to detect 1 case of cirrhosis. These numbers for family screening for HH are more favorable than numbers needed to detect a case in current population screening programs for breast, colon and cervical cancer, that is 1 in~1500–2000.133, 134, 135
Finally, we retrieved a study showing that greater awareness of HH and its potential consequences among probands and family members are important motivators for them to undergo screening for the disorder.136
In aggregate, there is sufficient evidence (i) to support screening for the p.C282Y variant in HFE in first-degree family members (siblings, children and parents) of clinically detected p.C282Y homozygotes or p.C282Y homozygotes detected otherwise, and (ii) to consider screening spouses when testing two or more children.
Follow-up of unaffected p.C282Y homozygotes or compound heterozygotes
It is recognized that early treatment to normalize iron stores increases the likelihood that the potential serious complications of the disease are effectively prevented, morbidity and mortality are reduced, and quality of life is increased.80, 122, 124, 131, 137, 138, 139 Moreover, it was recently shown in a cohort of 1086 p.C282Y homozygous patients treated at early stages that they have a long-term survival superior to the general population, with a decreased number of deaths by cardiovascular disease or extra-hepatic cancer,140 further supporting the benefit of sustained phlebotomy treatment of iron overload in HH. The question remains, however, as to whether preventive treatment should or not be recommended to all unaffected but genetically confirmed individuals because there is no evidence that all of these subjects will necessarily develop iron overload-related morbidity. To address this question, a systematic literature search was performed as described above. Search terms were phrased as PICO where patients (P) were defined as clinically or biochemically unaffected p.C282Y homozygotes or p.C282Y/p.H63D compound heterozygotes; interventions (I) were the follow up of iron indices (TS, SF) and phlebotomy treatment in comparison (C) with no intervention, and the expected outcome (O) was the evolution of iron indices and/or development of clinical symptoms. All systematic reviews or meta-analyses and comparative designs (cohort studies, case–control studies and cross-sectional studies) were eligible for inclusion. Case studies and case series with fewer than 20 patients were excluded. A total of 244 identified studies were first evaluated for further retrieval based on the question and a clear definition of the target population, focusing on population-based studies of unaffected subjects. The main reasons for exclusion were: (i) inappropriateness for the question, that is, associations of HFE genotypes as risk factors for other disease endpoints, studies in children, prevalence of HFE variants in other clinical settings or in case-finding studies and studies of genetic modifiers or (iii) possible bias or indirectness (populations highly selected for geographical region, blood donors or primary care-based). Of 27 papers kept for full revision, 10 reported screening studies in the normal population80, 88, 103, 141, 142, 143, 144, 145, 146 and have been already included in previous reviews or meta-analyses addressing the penetrance of HFE genotypes.28, 34, 118, 147, 148, 149 Four studies reported screening studies in elderly populations, all confirming the detection of previously undiagnosed p.C282Y homozygotes with biochemical evidence of iron overload but no significant morbidity.150, 151, 152, 153 Finally, and for the purpose of the specific outcome of this element (ie, the natural history of untreated HFE-related hemochromatosis), we selected only studies with long-term follow-up of untreated subjects. Only six studies fulfilled this criterion: one retrospective154 and two prospective123, 155 HH cohort studies of untreated cases; and two prospective population-based cohort studies; one in Denmark110 and one in Australia (results published in Gurrin et al111, 156and Allen et al157). The Copenhagen City Heart Study110 genotyped 9174 individuals and identified 23 p.C282Y homozygotes who had been followed for 25 years, none of them having developed clinically overt hemochromatosis. The authors conclude that p.C282Y homozygotes identified during population screening, and not because of clinically overt HH, at most need to be screened for manifestations of hemochromatosis every 10–20 years. The HealthIron study in Australia is a subsample of the Melbourne Collaborative Cohort Study-MCCS which enrolled 41–514 subjects aged 40–69 years from 1990 to 1994 with a comprehensive active follow-up from 2003 to 2007 (12 years follow-up). The results were described in three independent papers. In 2008, Gurrin and co-workers156 analyzed 86 untreated p.C282Y homozygotes and predicted the probability of SF and TS at follow-up exceeding the clinical thresholds from baseline under a multivariate normal model. They concluded that the probability of a baseline SF of 300–1000 ng/ml to progress to values higher than 1000 ng/ml is 13–35% in males and 16–22% in females. The probability of a normal baseline to progress for SF>1000 ng/ml if untreated is less than 15%. Finally, they concluded that the majority of p.C282Y homozygotes who are likely to develop a SF higher than 1000 ng/ml will have done so by the age of 55 years. The same authors also analyzed the evolution of iron indices in 180 compound heterozygotes for p.C282Y and p.H63D identified in the HealthIron study.111 They concluded that in these, the mean iron indices did not change during middle age in males but SF increases in females. Although maintaining elevated iron indices during middle age, documented iron overload-related disease in compound heterozygotes is rare. Finally, in 2010, Allen and co-workers157 analyzed the evolution of 102 untreated p.C282Y homozygotes and no evidence was found that those with SF concentrations below 1000 ng/ml at either baseline or 12 years later were at increased risk of HH-associated signs and symptoms. These results raise the question as to whether p.C282Y homozygotes with SF concentrations <1000 ng/ml should be managed aggressively or simply monitored to prevent SF rising over the critical threshold. Ideally, a randomized controlled trial of phlebotomy versus a ‘watchful waiting’ approach should be performed, but the time required to produce definitive results may be prohibitively long.
In conclusion, there is enough evidence to support the notion that iron indices may progress in some p.C282Y homozygotes or p.C282Y/p.H63D heterozygotes, supporting the recommendation for a biochemical follow-up. There is no evidence, however, to support a specific follow-up time or schedule. There is insufficient evidence to recommend phlebotomy treatment in unaffected (ie, without evidence of hemochromatosis) homozygotes or p.C282Y/p.H63D compound heterozygotes, but there is still a need for high-quality observational data on the treatment of HH.
The best practice meeting
In order to develop the initial guidelines draft into a consensus document, a workshop in the format of an EMQN Best Practice Meeting (BPM) was held in Porto, Portugal on 14 and 15 May 2014. The BPM was advertised using the same procedure as for the survey (see above) and this draft document, together with the topics for discussion, were made available before the BPM to the respondents who agreed to participate. The list of meeting attendees and respective affiliations is given in Supplementary Table 1.
As a result of the meeting, an agreed set of best practice guidelines was updated for genetic diagnostic and predictive testing for HFE-related HH and for reporting the results of such testing. In addition, some basic guidelines were also developed for testing and reporting the rarer forms of non-HFE-related HH. Resulting recommendations are based on available published evidence scored according to the GRADE system for quality and strength of evidence. If the quality of evidence was low or very low, the proposed recommendations resulted from the experts’ collective decision, this information being provided in the document. A final version of the consensus document (corrections and amendments performed according to the results of the BPM) was reviewed by the drafting team, circulated among the BPM participants for revision and further amendments and was finally approved by the EMQN board.
The BPM provided the opportunity not only to discuss the controversial topics previously reviewed through the systematic literature search and its implications for future research (described above), but also to discuss some other relevant topics described here.
Questions regarding nomenclature and case definition
It became apparent during the BPM that questions regarding the nomenclature and case definition of HH are among the major causes of confusion when dealing with the interpretation of test results and reporting practices. Some participants pointed to the fact that, in face of the increasing awareness and knowledge of the pathogenesis of iron overload disorders, some concepts relating to HH definition should be revised.
The term Hemochromatosis was first coined by von Recklinghausen158 in 1889 following the description of its iron-related pathology. The clinical characteristics of the disease were later described by John Sheldon159 in 1934 after a systematic review of autopsy cases, and the autosomal recessive mode of transmission was demonstrated by Simon and co-workers160 in 1977 following the discovery of its association to HLA. Since that time, a typical phenotypic characterization of patients was necessary to define HH, including the demonstration of severe liver iron overload. The selection of HH patients based on a strict phenotypic case definition was fundamental to permit the positional cloning of the HH-associated gene. After the discovery in 1996 of the genetic defect in HFE, which explained over 80% of previously phenotypically characterized cases,48 a genetic definition of HH became available, and a pre-symptomatic diagnosis in subjects with less severe forms of iron overload became feasible. These advances, however, also brought some confusion among clinicians about what constitutes the case definition of HH.27 Although a purely genetic definition cannot be accepted, owing to the low penetrance and variable clinical expression of p.C282Y homozygosity in HFE, a purely phenotypic case definition is also not acceptable because it will necessarily combine many different types of iron overload (genetic and acquired) defeating the original purpose of having a case definition, that is, to characterize the natural history, disease prognosis and, most importantly, to guide its treatment and/or prevention.27 For the purpose of nomenclature harmonization, we refer in this document to HFE-related HH according to the case definition agreed on the 2010 EASL Clinical Practice Guidelines for HFE Hemochromatosis, that is, as the occurrence of increased body iron stores associated with homozygosity for the p.C282Y HFE variant, with or without clinical symptoms.28 The other rare forms of HH are here classified as non-HFE-related HH in harmonization with the classification agreed in the AASLD Practice Guideline of 2011.4 They can be generally defined as increased body iron stores not associated with homozygosity for the p.C282Y HFE variant but attributed to pathogenic variants in other iron-related genes, namely hemojuvelin (HJV), hepcidin (HAMP) and transferrin receptor 2 (TFR2). For all of these genes, the pathogenesis of HH involves inadequate or ineffective production or hepcidin-mediated downregulation of ferroportin with consequent increase in iron absorption and iron export from macrophages. In the case of ferroportin variants, when they are associated with an excessive ferroportin-mediated iron export (gain-of-function variants, or type B), they cause a phenotype similar to the one observed in the classical HFE-related HH with elevated TS, hyperferritinemia and iron overload mostly affecting hepatocytes, but with normal or elevated (rather than low) hepcidin levels.25 In this case, ferroportin disease is included in the differential diagnosis of non-HFE-related hemochromatosis. In the case of the most common loss-of-function ferroportin variants (type A), the interaction between hepcidin and ferroportin is altered affecting iron export from cells with consequent accumulation in Kupffer cells and other macrophages, and is expressed by hyperferritinemia with high/normal or low TS.161 Although originally described as autosomal dominant hemochromatosis162 and referred to in the OMIM database as type 4 hemochromatosis, strictly speaking, it does not fit into the classical definition of HH which implies an inappropriately high absorption of iron by the gastrointestinal mucosa with consequent increased iron storage.
Controversy remains regarding the definition of HH associated with HFE variants other than p.C282Y, and no established consensus has been published before. The most frequently found HFE variant in all populations is p.H63D, and this was originally described by Feder and co-workers48 as a common variant associated with iron overload in patients who were p.C282Y heterozygotes. It should be noted that the two variants are always inherited in trans (on separate alleles) because they arose as independent founder alleles. After this original report, it was common practice to classify individuals with the p.C282Y and p.H63D variants inherited in compound heterozygosity as an HH-associated genotype. This practice, however, was later challenged by the growing evidence that p.C282Y/p.H63D compound heterozygous subjects have a distinct iron overload phenotype (mild to moderate) normally associated with the presence of other comorbidity factors, namely alcohol abuse or dysmetabolic iron overload syndrome.28, 111 Therefore, notwithstanding a role of p.H63D as a susceptibility factor for increased iron overload,118, 147, 163 for practical clinical purposes, p.C282Y/p.H63D compound heterozygotes and p.H63D homozygotes are not classified as HFE-associated HH (following the most recent EASL guidelines), and it is recommended that in patients with these genotypes and iron overload, other genetic or environmental risk factors should be examined.28 Another variant, p.S65C, reported as a normal variant in the European population,164 has also been described as a susceptibility allele in patients with iron overload, particularly in association with heavy alcohol consumption,146, 165 but no evidence has ever been provided to support a causative role in HH. Therefore, neither testing nor reporting the presence of p.S65C is recommended in the diagnosis of HFE-related HH. Some very rare HFE variants or deletions associated with disruption of the HFE protein function have been described in compound heterozygosity with p.C282Y in severe cases of HH.12 Although such patients can be classified as HFE-related HH, because of their rarity and necessary specific molecular workup for diagnosis, we suggest distinguishing these hemochromatosis variants from classical HFE-associated HH and including them in the classification of Rare Hereditary Hemochromatosis. Finally, it should be noted that in many clinical reports and disease databases, different nomenclatures have been used to classify HH and this fact may lead to confusion among clinicians and laboratory geneticists. A summary of the nomenclature used in this document, the disease names, synonyms, symbols, databases reference numbers (ORPHANET and OMIM (the Online Mendelian Inheritance of Man database)), the respective genes/loci and cytogenetic positions, as well as their major pathological and clinical defining characteristics, is given in Table 1.
The value of recognized Expert Centers
The French Reference Centre for Rare Iron Overload Diseases of Genetic Origin was created in 2007, as part of the first French National Plan for Rare Diseases (which started in 2005). Located at the University Hospital Pontchaillou in Rennes, and benefiting from a specific budget, it works in close collaboration with nine centers of competence spread across the country. The goal of this network of expertise is to contribute to the equity of medical care and research activities within the broad spectrum of rare diseases. The specific diseases involved correspond essentially to non HFE-related genetic iron overload diseases (type 2, 3 or 4 hemochromatosis), and to some other entities such as hereditary aceruloplasminemia and atransferrinemia. It also includes exceptional forms of HFE-related hemochromatosis involving rare variants. From the diagnostic viewpoint, the reference center analyzes all referred cases of unexplained iron excess using a multidisciplinary approach, in order to select those who warrant targeted genetic exploration. The center leads in proposing diagnostic and therapeutic recommendations to be disseminated to the medical community. With regard to basic and clinical research, the center proposes and coordinates various themes covering epidemiological, diagnostic and therapeutic aspects, in association with the recognized centers of competence. One of the hot topics today, considering increasingly efficient techniques in the field of molecular genetics, is to develop functional tests to aid in the prediction of deleterious effects associated with previously unclassified variants. A major aim is the integration, as soon as possible, of the reference center within a European network for rational sharing of diagnostic tools, homogenization of diagnostic and therapeutic practices, and coordination of research actions. There remains a great disparity in Europe regarding the policy on rare diseases, from countries that have recently decided to develop a plan/strategy to those whose national plans and strategies are fully implemented. However, many European medical, scientific and patient organizations are actively engaged in this cause, which gives confidence in future improvement of the management of rare diseases, including those related to genetic iron overload.
The future of NGS in clinical practice
Historically, genetic testing for HH has developed from detection of the common HFE variants through to conventional DNA sequencing of the HFE gene, and/or other genes implicated in rarer hereditary iron overload disorders. With the advent of NGS, it is now possible to investigate, on a research basis, the genetics of rare or complex disorders, based on whole-exome or -genome analyses. The technology for sequencing multiple genes simultaneously is developing rapidly, with no firm consensus on approaches to diagnostic testing. However, guidance on consensus standards for identifying and reporting variants identified using NGS approaches has been produced.166 In a diagnostic context, NGS based on targeted gene analysis is being introduced, including panels linked to disorders of iron metabolism. For routine type 1 HH analysis the current approach, based on recognized phenotypic parameters to identify index cases that have a phenotype meriting p.C282Y genotyping prior to relevant predictive or diagnostic testing in appropriate family members, is likely to remain in place for the foreseeable future. Where NGS gene panel analysis will increasingly have a role is in those rare cases where there is documented evidence of iron overload and initial screening approaches do not detect recognized variants or variants associated with the phenotype. This will apply not only to type 1 HH but also to the other forms discussed in this document. In such circumstances, iron metabolism gene panels can be applied to facilitate detection of rare variants, ultimately contributing to our understanding of these disorders. For this strategy to be successful, such investigations will be best performed by referral to a small number of specialist centers in order to ensure both a cost-effective approach and the availability of the necessary expertise in the interpretation of results obtained, particularly when interpreting the likely significance of any unclassified variants detected.
A final remark: the patients’ expectations and the role of General Practitioners in the management of HH
In the frame of the present guidelines, a survey was also conducted by the European Federation of Associations of Patients with Hemochromatosis (EFAPH) on the perceptions of patients about genetic information namely on the sources used or trusted. The results of the survey provided, for the first time, a basis for recommendations intended to improve communication between patients and health-care providers regarding genetic information and suggests that more efforts should be put in awareness, motivation and education among General Practitioners regarding HH management.167
The guidelines—evidence-based recommendations
These guidelines offer an overview of the recommended basis for genetic testing referral where there is a suspicion of type 1 HH or rare forms of HFE-related and non-HFE-related HH, along with predictive testing of adult first-degree relatives of subjects confirmed to have HH by molecular analysis. They illustrate the minimum expected interpretation standards for these disorders where the correlation between the clinical phenotype and genotype must be considered prior to referral for investigation and reporting.
Methods and testing strategies
A variety of methods for HFE genotyping are in use by laboratories participating in the EMQN EQA scheme for HFE HH. The most common testing strategies in use detect the p.C282Y (c.845G>A) and p.H63D (c.187C>G) variants and are based on PCR/restriction enzyme digestion or various allele-specific amplification strategies.
The vast majority of HFE-related hemochromatosis is associated with homozygosity for p.C282Y. p.C282Y/p.H63D compound heterozygosity may be a risk factor predisposing to mild or moderate forms of iron overload when in association with comorbidity factors, for example, alcohol or metabolic syndrome.3, 111 The association of homozygosity for p.H63D with iron overload is debated and requires further clinical research, but it is now recommended that other risk factors or other genetic causes should be sought and investigated in patients with this genotype and demonstrated iron overload.3 Testing for these two HFE variants can be performed either sequentially or simultaneously. Sequential testing, starting with p.C282Y, will avoid detection of p.H63D homozygotes, and this has been considered by most experts as an advantage. However, this reflex testing approach increases the turn-around time and overall cost. A simultaneous testing strategy should be weighed against the possibility of creating anxiety or unnecessary investigation. The laboratory should take these factors into account when deciding to use this strategy. In any case, when a method detects both variants concurrently, which is generally the case, there is an obligation to report the complete result.
There is no supporting evidence for a role of the p.S65C variant (c.193A>T) in clinically manifest HH (p.S65C is a normal variant in the general European population).146, 164, 168 Therefore, testing for the p.S65C variant is not recommended for diagnostic purposes; if detected as an incidental finding, it should not be reported to avoid possible over-interpretation of its significance.
Guidelines from the European Society of Human Genetics and the American College of Medical Genetics recommend not testing minors for carrier status for late-onset disorders. When requested by parents in this situation, local and international practice and regulations should be followed/recommended by the health careers.
Quality assurance framework
The EMQN external quality assessment (EQA) scheme for HFE continues to detect both genotyping errors and variable quality of interpretive comments. This indicates the continued importance of quality assurance and improvement. It is recommended that testing laboratories are accredited to international standards (ISO 15189 or equivalent).169, 170 The techniques used should be validated in-house with appropriate sample types and numbers. If CE-marked IVD kits are used, in-house verification should be performed. Laboratories reporting HFE testing results should participate annually in EQA. EQA provides a long-term, retrospective assessment of laboratory performance, allowing laboratories to demonstrate consensus with their peers and providing information on inter-method comparability. Unless dictated by legislation, the choice of EQA provider lies with the laboratory, but the use of an EQA programme that assesses reporting as well as genotyping and that is accredited to ISO 17043 is recommended, wherever possible.
Recommendations for HFE testing strategies:
-
Laboratories providing testing for HFE-associated HH should test for p.C282Y (1A).
-
According to local practice, p.H63D can be a considered an optional complementary test that can be offered sequentially or simultaneously to p.C282Y testing (2C).
-
Testing for p.S65C should not be offered; if detected as an incidental finding, it should not be reported (1B).
-
It is recommended that testing laboratories are accredited according to international standards (ISO 15189 or equivalent).
-
It is recommended that genetic testing for suspected rare forms of hemochromatosis should be performed by specialist centers.
Criteria for diagnostic and predictive testing
These have been extensively covered in published clinical guidelines and reports (summarized in 'Results of the survey on current practices of genetic diagnosis of HH', see Swinkels et al9and Berwouts et al170). In current clinical practice, patients are often referred for HH genetic testing based on a diverse range of indicators. These include isolated raised SF (hyperferritinemia), raised transaminases, type 2 diabetes and arthralgia. This situation has arisen owing to overestimations of the penetrance and clinical impact of p.C282Y homozygosity and these patient groups are no longer considered to be appropriate for HFE-HH genetic testing. In the absence of raised TS, there is no evidence to support genetic testing for the p.C282Y variant in an index case solely for clinical reasons.
Given the importance of the TS result, it should be noted that the concept of ‘elevated transferrin saturation’ varies, ranging from levels of 45 to 60%. A higher cutoff value is expected to have a superior positive predictive value and is useful in populations where reference values are higher, usually associated with lifestyle habits including regular daily alcohol consumption.29 Lower cutoff values are more sensitive (with a higher negative predictive value), but less specific. They may facilitate earlier detection of iron overload but will increase false-positive detection rates.171 Elevated SF (ie, exceeding the local upper limit of the reference range) is a potential biochemical marker of iron overload but is not specific for HH. Therefore, confirmed hyperferritinemia should prompt testing for TS before genetic testing. If TS is normal, alternative causes of hyperferritinemia should be sought, such as indicators of inflammation and liver disease, most commonly observed in the presence of metabolic syndrome or fatty liver disease.31 Conversely, in the presence of high TS with normal SF values, a genetic test should be considered. Testing for rare variants in HFE, or other genes associated with non-HFE-related HH, should be reserved for highly selected cases with documented, unexplained iron overload (increased body iron stores) that test negative for p.C282Y homozygosity. These rare cases should be referred to specialist centers offering appropriate molecular diagnosis and expert advice on iron overload disorders.
Predictive testing of first-degree relatives of subjects confirmed to have HH by molecular analysis
Because the prevalence of individuals at risk to develop morbidity is increased among first-degree relatives of index cases, their identification through predictive testing is appropriate. Genetic testing of adult asymptomatic individuals should be undertaken only after appropriate counselling addressing the pros and cons of testing as well as the possible clinical consequences relating to the test result.
p.C282Y homozygotes
Genetic testing of adult first-degree relatives of p.C282Y homozygous cases aims to identify covert homozygotes in the family so that appropriate management can then be instigated. The low penetrance of p.C282Y homozygosity should be clearly explained before testing. When a p.C282Y heterozygote is identified by this process, further cascade testing of his first-degree relatives is not recommended. This rationale is based on low morbidity when the further reduced risk for inheritance of p.C282Y homozygosity in these individuals is combined with the low penetrance of the clinical phenotype of HH in homozygotes.
p.C282Y/p.H63D compound heterozygotes
Owing to the relatively low risk of morbidity and the consequently low clinical efficiency of screening, consensus was not reached in order to recommend genetic testing of clinically asymptomatic adult first-degree relatives of p.C282Y/p.H63D compound heterozygotes.
Other HFE genotypes
Owing to the relatively low risk of morbidity and the low penetrance, genetic testing of clinically asymptomatic adult first-degree relatives of p.C282Y heterozygotes or p.H63D homozygotes is not recommended.
Rare forms of HFE- or non-HF- related HH
Genetic testing of clinically asymptomatic adult first-degree relatives of individuals with these rare conditions aims to identify additional individuals at risk of these rare forms of HH and it is therefore recommended.
Recommendations for diagnostic and predictive testing:
-
Population screening for the p.C282Y variant is not currently recommended (1B).
-
It is considered to be good practice to confirm elevated TS before HFE genetic diagnosis testing (1B).
-
Testing adult siblings (brothers and sisters) of p.C282Y homozygotes is recommended owing to the increased risk of p.C282Y homozygosity and related increased morbidity (1B).
-
Testing adult offspring of p.C282Y homozygotes is recommended owing to increased risk of p.C282Y homozygosity and related increased morbidity (1C).
-
Testing asymptomatic parents of p.C282Y homozygotes is not recommended systematically but rather as a clinical decision depending on their age, sex and ferritin, all three influencing the probability to develop severe iron overload (1C).
-
Systematic testing of adult first-degree relatives of p.C282Y heterozygotes is not currently recommended, in the absence of evidence of benefit (2C).
-
HFE testing of minors is not recommended (1B).
-
Prenatal diagnosis is not appropriate in HFE-related HH because it is a treatable, adult onset condition (1C).
-
Genetic testing of first-degree relatives of rare HFE-related and non-HFE related HH should be performed by specialist/reference centers/laboratories (2C).
Reporting of HFE genotyping results
The primary role for HFE genetic testing is to confirm or exclude a diagnosis of HFE-related HH, or to predict the risk for this condition. No solid evidence has been provided to support the clinical utility of HFE genotyping as a susceptibility test for iron overload in general. Analysis of current practice in HFE molecular testing reveals that a growing number of clinicians routinely request this genetic test but that it confirms a diagnosis of HFE-related HH in only a small proportion of cases.1 If not properly informed as to the relative significance of each genotype combination, it is possible that some clinicians may incorrectly assume a diagnosis of HH for genotypes other than p.C282Y homozygosity, with undesirable consequences relating to both patient management and counselling information provided to relevant family members. It is therefore recommended that when reporting HFE genotyping results, a clear statement be given regarding the requested reason for testing, and that interpretative comments are given in a case-specific context. This implies, of course, that a clear indication of the reason for testing is provided by the requesting clinician.
Additional interpretative comments on reports may refer to: the implications of the test result for other family members, the need to establish differential diagnosis with other forms of iron overload and suggested follow-up strategies in the case of predictive detection. For example, the identification of an asymptomatic or previously undetected p.C282Y homozygous family member should prompt evaluation of their iron indices and establishment of an appropriate follow-up program. If asymptomatic p.C282Y/p.H63D compound heterozygotes are detected, recommendations regarding monitoring of iron indices are more controversial and a consensus was not reached yet. The fact that individuals with this genotype may go on to exhibit a mild-to-moderate iron overload phenotype, normally associated with other comorbidity factors such as alcohol abuse or metabolic syndrome,3, 111 may constitute an argument to recommend referral to a specialist for monitoring of iron indices and lifestyle habits. Nevertheless, care should be taken not to create unnecessary anxiety in apparently healthy subjects with a compound heterozygous genotype. Although they might have elevated iron indices, documented iron overload-related disease in subjects with this genotype is rare.111
Reporting scenarios
The following provides basic guidelines for reporting HFE test results based on reasons for referral and resulting genotypes. Points regarded as essential are highlighted in italics. It should be stressed that these guidelines are presented under the assumption that HFE genotyping is performed to confirm, exclude or predict the most common HFE-related forms of HH and they do not cover rare or population-specific alternative variants in HFE. Other forms of iron overload and other types of HH exist and further investigation may be suggested where relevant clinical and biochemical features indicate. In those rare cases with a strongly suggestive phenotype that are not confirmed to be p.C282Y homozygous, investigation for other genetic forms of HH or referral to a specialist unit may be suggested. However, it must be stressed that these are rare and should only be considered in cases where there is a documented severe iron overload that remains unexplained after exclusion of other causes. Reference to the implications of the HFE test result for other adult family members and recommendations for genetic counselling should be reserved only for individuals carrying the p.C282Y variant; otherwise it may create unnecessary anxiety. In cases where screening is performed with additional genetic and/or phenotypic data, reporting recommendations may be adapted to reflect individual case scenarios more closely. A summary of basic interpretive comments relating to HFE-related HH is given in Table 2.
Diagnostic reporting scenarios
Diagnostic referral (ie, affected individual); homozygous p.C282Y. The individual is homozygous for the p.Cys282Tyr variant in the HFE gene.
Reports should state that, in the presence of a suggestive phenotype, this genotype supports the diagnosis of HFE-related HH. Formal diagnosis requires demonstration of increased hepatic iron stores.
Additional comments may refer to implications of the result for other adult first-degree family members and recommend genetic counselling for predictive/carrier testing. It is not appropriate to simply state that all relatives must/should be tested. Genetic testing for p.C282Y and biochemical testing for iron overload is recommended in first-degree adult family members.
Diagnostic referral (ie, affected individual); compound heterozygous p.C282Y/p.H63D C282Y. Patients with this genotype may have iron overload but to a much lesser degree than p.C282Y homozygous HH patients, and usually in association with other risk factors.111 Although compound heterozygotes might maintain elevated iron indices during middle age, documented iron overload-related disease is rare and this genotype should therefore have a different interpretation than for a p.C282Y homozygote referred on the same basis. The genotype is common among the general European population and a reference to the local estimated frequency may be given.
The individual is a carrier of both the p.Cys282Tyr and the p.His63Asp variants in the HFE gene.
Reports should state that the diagnosis of the most common HFE-related HH is excluded. This genotype may predispose to mild / moderate iron overload. In patients with iron overload, other contributing factors should be considered (most commonly, alcohol consumption, fatty liver disease, and/or metabolic syndrome).
In patients with a severe iron overload phenotype, other rare forms of HH cannot be excluded.
Additional comments may refer to implications of the result for other adult first-degree family members, and genetic counselling may be considered. It is not considered appropriate to recommend predictive/carrier testing to all relatives (cascade family screening). Biochemical testing of first-degree adult family members should be considered.
Diagnostic referral (ie, affected individual); heterozygous p.C282Y. It has been documented that some p.C282Y heterozygotes may exhibit mild to moderately raised indices of iron overload,106 although complications due to iron overload are very rare and may be influenced by additional factors, both genetic and environmental.
The individual is a carrier of the p.Cys282Tyr variant in the HFE gene.
Reports should state that the diagnosis of the most common HFE-related HH is excluded.
In patients with a severe iron overload phenotype, other rare forms of HH cannot be excluded.
Additional comments may refer to implications of the result for other adult first-degree family members, and genetic counselling may be considered. It is not considered appropriate to recommend predictive/carrier testing to all relatives (cascade family screening). Biochemical testing of first-degree adult family members may be considered.
Diagnostic referral (ie, affected individual); homozygous p.H63D. It has been suggested that p.H63D homozygotes may have a slight risk of iron overload.118, 147, 163 Despite some geographical variation118 this genotype is present in about 3% of the general European population and its significance as a susceptibility factor remains uncertain.
The individual is homozygous for the p.His63Asp variant in the HFE gene.
Reports should state that the diagnosis of the most common HFE-related HH is excluded. This genotype may be associated with a slight increase of ferritin and TS. Other causes of iron overload should be considered.
In patients with a severe iron overload phenotype, other rare forms of HH cannot be excluded.
Additional comments may refer to the uncertainty and controversy regarding the role of p.H63D as a risk factor for mild to moderate iron overload, in the presence of other causes of iron overload (eg, fatty liver disease and/or metabolic syndrome). It is not appropriate to refer to implications of this genotype for other adult family members.
Diagnostic referral (ie, affected individual); heterozygous p.H63D. The individual is a carrier of the p.His63Asp variant in the HFE gene. This variant is present at a high frequency in the general population. The diagnosis of the most common HFE-related HH is excluded and other causes of iron overload should be considered.
In patients with a severe iron overload phenotype, other rare forms of HH cannot be excluded.
It is not appropriate to refer to implications of this genotype for other family members.
Diagnostic referral (ie, affected individual); detection of the p.S65C variant. Given the absence of any clinical merit, testing for p.S65C is not recommended. If p.S65C is detected by the analysis method in use, this variant should not be reported. Therefore, report wording should treat such diagnostic cases as ‘p.C282Y and p.H63D variants not detected’ (see below).
Diagnostic referral (ie, affected individual); p.C282Y and p.H63D variants not detected. Reports should state that the diagnosis of the most common HFE-related HH is excluded and other causes of iron overload should be considered.
In patients with a severe iron overload phenotype, other rare forms of HH cannot be excluded.
Predictive reporting scenarios
The guidance for reporting predictive/carrier testing results are written under the assumption that the iron status of the individual is not known and that appropriate genetic counselling has been performed prior to testing.
In cases where screening is performed with additional genetic and/or phenotypic data, reporting recommendations may be adapted to reflect individual case scenarios more closely.
Predictive referral (ie, individual currently unaffected); homozygous p.C282Y. The individual is homozygous for the p.Cys282Tyr variant in the HFE gene. He is at an increased risk of developing HFE-related HH, and it is recommended that serum iron parameters (TS and ferritin) be monitored.
The report may refer to implications of the result for other adult family members and suggest that genetic counselling be considered if not previously performed. Given the variable penetrance of p.C282Y homozygosity, it is not considered appropriate to state that the individual has, or will go on to develop, HH if no evidence of phenotypic expression is given.
Predictive referral (ie, individual currently unaffected); compound heterozygous p.C282Y/p.H63D. The individual is a carrier of both the p.Cys282Tyr and p.His63Asp variants in the HFE gene and is at low risk of developing significant iron overload. He may be at risk of developing mild-to-moderate iron overload if there is an association with other comorbidity factors (eg, alcohol abuse, fatty liver disease and/or metabolic syndrome).
Additional comments may suggest monitoring of iron indices.
It is not considered appropriate to recommend predictive/carrier testing to relatives.
Predictive referral (ie, individual currently unaffected); all other non-p.C282Y-carrying genotypes. The individual is not a carrier of the p.Cys282Tyr variant in the HFE gene and he is at no increased risk of developing HFE-related HH.
Predictive referral (ie, individual currently unaffected); heterozygous p.C282Y. The individual is a carrier of the p.Cys282Tyr variant in the HFE gene and he is at no increased risk of developing HFE-related HH.
It is not considered appropriate to recommend predictive/carrier testing to relatives.
Reporting results for rare forms of HFE- or non-HFE-related HH
General information on requirements for variant reporting can be found in appropriate nomenclature guidelines. It is recommended that Human Genome Variation Society (HGVS) convention is followed when reporting variants and unclassified variants.172, 173 Information on previously described variants can be found in available databases,174 however, the creation of locus-specific databases listing pathogenic and non-pathogenic variants is encouraged. The following represents some basic guidelines for reporting test results based on possible scenarios.
Reporting on a known causative variant in the tested genes (HFE2 (HJV), HAMP, TFR2 or SLC40A1) in an affected index case
The report should state that a causative variant(s) has been detected and that this confirms the clinical diagnosis.
The report should include a description of the reason why a particular variant(s) is considered causative, drawing on all relevant supporting data including the scientific published data.
Determination of the parental origin of the detected variant(s) is advised.
Predictive/carrier testing of relatives at risk should be offered after appropriate genetic counselling.
Ferroportin disease exhibits autosomal dominant inheritance associated with, in its usual form (type A), hepatic and splenic iron overload in the absence of increased TS. In the type B form, its phenotype mimics that of classical HH. Heterozygous SLC40A1 variants may be sufficient to cause these particular phenotypes and this should be taken into account when conducting family screening.
Reporting on a novel variant in the tested genes (HFE, HFE2 [HJV], HAMP, TFR2 or SLC40A1) in an affected index case
If the genetic testing procedure has identified an unclassified variant as the only sequence change, this should be interpreted according to reporting of unclassified variants guidelines.173
General guidelines for reporting
General information on requirements for variant reporting can be found in ISO 15189,170 the OECD Guidelines for Quality Assurance in Molecular Genetic Testing and the Swiss Medical Genetics Society guidelines for reporting.38, 175
General report contents
According to the international standards for accreditation, reports should be written in a clear, concise, accurate, fully interpretative, credible and authoritative format. Importantly, they should specify the scope of the investigation, that is, if testing is for HFE-related HH, or if the rarer forms are being considered.
Nomenclature
It is considered essential that reports follow HGVS guidelines for reporting variants.172 Use of HGVS requires coding changes at the nucleotide level and citation of the appropriate reference sequence. In addition, protein-based coding is permissible (three-letter). However, this should not be at the expense of a clear and succinct description of the genotype. Where both common and formal names for variants are used, these should be formatted in such a way as to avoid confusion to the report recipient.
The correct HGVS coding of the principal genotypes is given in Table 2. HGVS nomenclature can be checked using the freely-accessible program Mutalyzer.176
Conclusion
An agreed set of best practice guidelines has now been updated for diagnostic, predictive and carrier testing for HFE-related HH and for reporting the results of such testing. In addition, some basic guidelines have been developed for testing and reporting rarer forms of non-HFE-related HH. Consensus and consultation processes for this document occurred within the framework of a best practice meeting where, following prior dissemination of related literature, relevant topics were discussed by a group of experts. The resulting recommendations are based on the available published evidence, and the GRADE system for scoring the quality of evidence and strength of recommendations was applied.36 This process has been translated into guidance when reporting variants associated with iron overload disorders.
References
Fleming RE, Ponka P : Iron overload in human disease. N Engl J Med 2012; 366: 348–359.
Mayr R, Janecke AR, Schranz M et al: Ferroportin disease: a systematic meta-analysis of clinical and molecular findings. J Hepatol 2010; 53: 941–949.
European Association For The Study Of The Liver: EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol 2010; 53: 3–22.
Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS : Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54: 328–343.
van Bokhoven MA, van Deursen CT, Swinkels DW : Diagnosis and management of hereditary haemochromatosis. BMJ 2011; 342: c7251.
Brissot P : Diagnosis and current treatments for primary iron overload. Am J Hematol 2007; 82: 1140–1141.
Adams PC, Barton JC : How I treat hemochromatosis. Blood 2010; 116: 317–325.
Zoller H, Cox TM : Hemochromatosis: genetic testing and clinical practice. Clin Gastroenterol Hepatol 2005; 3: 945–958.
Swinkels DW, Jorna AT, Raymakers RA : Synopsis of the Dutch multidisciplinary guideline for the diagnosis and treatment of hereditary haemochromatosis. Neth J Med 2007; 65: 452–455.
Camaschella C, Poggiali E : Rare types of genetic hemochromatosis. Acta Haematol 2009; 122: 140–145.
Brissot P, Bardou-Jacquet E, Jouanolle AM, Loreal O : Iron disorders of genetic origin: a changing world. Trends Mol Med 2011; 17: 707–713.
Aguilar-Martinez P, Grandchamp B, Cunat S et al: Iron overload in HFE C282Y heterozygotes at first genetic testing: a strategy for identifying rare HFE variants. Haematologica 2011; 96: 507–514.
Le Gac G, Congiu R, Gourlaouen I, Cau M, Ferec C, Melis MA : Homozygous deletion of HFE is the common cause of hemochromatosis in Sardinia. Haematologica 2010; 95: 685–687.
Lok CY, Merryweather-Clarke AT, Viprakasit V et al: Iron overload in the Asian community. Blood 2009; 114: 20–25.
Hayashi H, Wakusawa S, Motonishi S et al: Genetic background of primary iron overload syndromes in Japan. Intern Med 2006; 45: 1107–1111.
McDonald CJ, Wallace DF, Crawford DH, Subramaniam VN : Iron storage disease in Asia-Pacific populations: the importance of non-HFE mutations. J Gastroenterol Hepatol 2013; 28: 1087–1094.
Kato J, Fujikawa K, Kanda M et al: A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am J Hum Genet 2001; 69: 191–197.
Andriopoulos B Jr, Corradini E, Xia Y et al: BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet 2009; 41: 482–487.
Vujic Spasic M, Sparla R, Mleczko-Sanecka K et al: Smad6 and Smad7 are co-regulated with hepcidin in mouse models of iron overload. Biochim Biophys Acta 2013; 1832: 76–84.
Donker AE, Raymakers RA, Vlasveld LT et al: Practice guidelines for the diagnosis and management of microcytic anemias due to genetic disorders of iron metabolism or heme synthesis. Blood 2014; 123: 3873–3886, quiz 4005.
Camaschella C, Roetto A, De Gobbi M : Juvenile hemochromatosis. Semin Hematol 2002; 39: 242–248.
Bardou-Jacquet E, Cunat S, Beaumont-Epinette MP et al: Variable age of onset and clinical severity in transferrin receptor 2 related haemochromatosis: novel observations. Br J Haematol 2013; 162: 278–281.
Camaschella C, Roetto A, Cali A et al: The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet 2000; 25: 14–15.
Swinkels DW, Venselaar H, Wiegerinck ET et al: A novel (Leu183Pro-)mutation in the HFE-gene co-inherited with the Cys282Tyr mutation in two unrelated Dutch hemochromatosis patients. Blood Cells Mol Dis 2008; 40: 334–338.
Drakesmith H, Schimanski LM, Ormerod E et al: Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood 2005; 106: 1092–1097.
Deugnier Y, Turlin B : Pathology of hepatic iron overload. World J Gastroenterol 2007; 13: 4755–4760.
Adams PC : Hemochromatosis case definition: out of focus? Nat Clin Pract Gastroenterol Hepatol 2006; 3: 178–179.
European Association For The Study Of The Liver: EASLD clinical practice guidelines for HFE hemochromatosis. J Hepatol 2010; 53: 3–22.
Porto G, Vicente C, Fraga J, da Silva BM, de Sousa M : Importance of establishing appropriate local reference values for the screening of hemochromatosis: a study of three different control populations and 136 hemochromatosis family members. Hemochromatosis Clinical and Research Group. J Lab Clin Med 1992; 119: 295–305.
Adams PC, Speechley M, Barton JC, McLaren CE, McLaren GD, Eckfeldt JH : Probability of C282Y homozygosity decreases as liver transaminase activities increase in participants with hyperferritinemia in the hemochromatosis and iron overload screening study. Hepatology 2012; 55: 1722–1726.
Adams PC, Barton JC : A diagnostic approach to hyperferritinemia with a non-elevated transferrin saturation. J Hepatol 2011; 55: 453–458.
El-Serag HB, Inadomi JM, Kowdley KV : Screening for hereditary hemochromatosis in siblings and children of affected patients. A cost-effectiveness analysis. Ann Intern Med 2000; 132: 261–269.
Bulaj ZJ, Ajioka RS, Phillips JD et al: Disease-related conditions in relatives of patients with hemochromatosis. N Engl J Med 2000; 343: 1529–1535.
Whitlock EP, Garlitz BA, Harris EL, Beil TL, Smith PR : Screening for hereditary hemochromatosis: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med 2006; 145: 209–223.
Grosse SD, Rogowski WH, Ross LF, Cornel MC, Dondorp WJ, Khoury MJ : Population screening for genetic disorders in the 21st century: evidence, economics, and ethics. Public Health Genomics 2010; 13: 106–115.
GRADE Working Group http://www.gradeworkinggroup.org/index.htm.
EMQN Quality in the world of Molecular Genetics http://www.cmgs.org/BPGs/Best_Practice_Guidelines.htm.
Swiss Society of Medical Genetics http://www.sgmg.ch/view_page_professional.php?view=page&page_id=18.
Deutsche Gesellschaft für Humangenetik http://www.gfhev.de/en/documents/index.htm.
The European Molecular Genetics Quality Network http://www.emqn.org.
Eurogentest http://www.eurogentest.org/documents/.
Centre de référence des surcharges en fer rare d'origine génétique http://www.centre-reference-fer-rennes.org/.
Jouanolle AM, Gerolami V, Ged C et al: [Molecular diagnosis of HFE mutations in routine laboratories. Results of a survey from reference laboratories in France]. Ann Biol Clin 2012; 70: 305–313.
Haute Autorité de Santé http://www.has-sante.fr/portail/upload/docs/application/pdf/haemochromatosis_abstract.pdf.
British Committee for Standards in Haematology http://www.bcshguidelines.com/documents/haemochromotosis_2000.pdf, 2000.
Leitlinien zur molekular genetischen Diagnostik der HH http://www.gfhev.de/de/leitlinien/LL_und_Stellungnahmen/ 2008-12-Haemochromatose.pdf, 2008.
King C, Barton DE : Best practice guidelines for the molecular genetic diagnosis of Type 1 (HFE-related) hereditary haemochromatosis. BMC Med Genet 2006; 7: 81.
Feder JN, Gnirke A, Thomas W et al: A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996; 13: 399–408.
Bacon BR, Olynyk JK, Brunt EM, Britton RS, Wolff RK : HFE genotype in patients with hemochromatosis and other liver diseases. Ann Intern Med 1999; 130: 953–962.
Bauduer F, Scribans C, Degioanni A, Renoux M, Dutour O : Distribution of the C282Y and H63D polymorphisms in hereditary hemochromatosis patients from the French Basque Country. Ann Hematol 2005; 84: 99–102.
Bell H, Berg JP, Undlien DE et al: The clinical expression of hemochromatosis in Oslo, Norway. Excessive oral iron intake may lead to secondary hemochromatosis even in HFE C282Y mutation negative subjects. Scand J Gastroenterol 2000; 35: 1301–1307.
Beutler E, Gelbart T, West C et al: Mutation analysis in hereditary hemochromatosis. Blood Cells Mol Dis 1996; 22: 187–194, discussion 194a–194b.
Borot N, Roth M, Malfroy L et al: Mutations in the MHC class I-like candidate gene for hemochromatosis in French patients. Immunogenetics 1997; 45: 320–324.
Brandhagen DJ, Fairbanks VF, Baldus WP et al: Prevalence and clinical significance of HFE gene mutations in patients with iron overload. Am J Gastroenterol 2000; 95: 2910–2914.
Brissot P, Moirand R, Jouanolle AM et al: A genotypic study of 217 unrelated probands diagnosed as "genetic hemochromatosis" on "classical" phenotypic criteria. J Hepatol 1999; 30: 588–593.
Burt MJ, Upton JD, Morison IM, Chapman BA, Faed JM, George PM : Molecular analysis of HLA-H gene mutations in New Zealand patients with haemochromatosis. N Z Med J 1997; 110: 429–432.
Cardoso EM, Stal P, Hagen K et al: HFE mutations in patients with hereditary haemochromatosis in Sweden. J Intern Med 1998; 243: 203–208.
Carella M, D'Ambrosio L, Totaro A et al: Mutation analysis of the HLA-H gene in Italian hemochromatosis patients. Am J Hum Genet 1997; 60: 828–832.
Consortium U: A simple genetic test identifies 90% of UK patients with haemochromatosis. The UK Haemochromatosis Consortium. Gut 1997; 41: 841–844.
Datz C, Lalloz MR, Vogel W et al: Predominance of the HLA-H Cys282Tyr mutation in Austrian patients with genetic haemochromatosis. J Hepatol 1997; 27: 773–779.
De JD, Reta A, Castiella A, Pozueta J, Prada A, Cuadrado E : HFE gene mutations analysis in Basque hereditary haemochromatosis patients and controls. Eur J Hum Genet 2001; 9: 961–964.
De Marco F, Liguori R, Giardina MG et al: High prevalence of non-HFE gene-associated haemochromatosis in patients from southern Italy. Clin Chem Lab Med 2004; 42: 17–24.
Guix P, Picornell A, Parera M et al: Distribution of HFE C282Y and H63D mutations in the Balearic Islands (NE Spain). Clin Genet 2002; 61: 43–48.
Hellerbrand C, Bosserhoff AK, Seegers S et al: Mutation analysis of the HFE gene in German hemochromatosis patients and controls using automated SSCP-based capillary electrophoresis and a new PCR-ELISA technique. Scand J Gastroenterol 2001; 36: 1211–1216.
Jazwinska EC, Cullen LM, Busfield F et al: Haemochromatosis and HLA-H. Nat Genet 1996; 14: 249–251.
Jouanolle AM, Gandon G, Jezequel P et al: Haemochromatosis and HLA-H. Nat Genet 1996; 14: 251–252.
Moirand R, Jouanolle AM, Brissot P, Le Gall JY, David V, Deugnier Y : Phenotypic expression of HFE mutations: a French study of 1110 unrelated iron-overloaded patients and relatives. Gastroenterology 1999; 116: 372–377.
Mura C, Raguenes O, Ferec C : HFE mutations analysis in 711 hemochromatosis probands: evidence for S65C implication in mild form of hemochromatosis. Blood 1999; 93: 2502–2505.
Murphy S, Curran MD, McDougall N, Callender ME, O'Brien CJ, Middleton D : High incidence of the Cys 282 Tyr mutation in the HFE gene in the Irish population—implications for haemochromatosis. Tissue Antigens 1998; 52: 484–488.
Nielsen P, Carpinteiro S, Fischer R, Cabeda JM, Porto G, Gabbe EE : Prevalence of the C282Y and H63D mutations in the HFE gene in patients with hereditary haemochromatosis and in control subjects from Northern Germany. Br J Haematol 1998; 103: 842–845.
Piperno A, Sampietro M, Pietrangelo A et al: Heterogeneity of hemochromatosis in Italy. Gastroenterology 1998; 114: 996–1002.
Rivard SR, Mura C, Simard H et al: Mutation analysis in the HFE gene in patients with hereditary haemochromatosis in Saguenay-Lac-Saint-Jean (Quebec, Canada). Br J Haematol 2000; 108: 854–858.
Ryan E, O'Keane C, Crowe J : Hemochromatosis in Ireland and HFE. Blood Cells Mol Dis 1998; 24: 428–432.
Sanchez M, Bruguera M, Bosch J, Rodes J, Ballesta F, Oliva R : Prevalence of the Cys282Tyr and His63Asp HFE gene mutations in Spanish patients with hereditary hemochromatosis and in controls. J Hepatol 1998; 29: 725–728.
Sham RL, Ou CY, Cappuccio J, Braggins C, Dunnigan K, Phatak PD : Correlation between genotype and phenotype in hereditary hemochromatosis: analysis of 61 cases. Blood Cells Mol Dis 1997; 23: 314–320.
Sham RL, Raubertas RF, Braggins C, Cappuccio J, Gallagher M, Phatak PD : Asymptomatic hemochromatosis subjects: genotypic and phenotypic profiles. Blood 2000; 96: 3707–3711.
Van Vlierberghe H, Messiaen L, Hautekeete M, De Paepe A, Elewaut A : Prevalence of the Cys282Tyr and His63Asp mutation in Flemish patients with hereditary hemochromatosis. Acta Gastroenterol Belg 2000; 63: 250–253.
Willis G, Jennings BA, Goodman E, Fellows IW, Wimperis JZ : A high prevalence of HLA-H 845A mutations in hemochromatosis patients and the normal population in eastern England. Blood Cells Mol Dis 1997; 23: 288–291.
Adams PC, Kertesz AE, McLaren CE, Barr R, Bamford A, Chakrabarti S : Population screening for hemochromatosis: a comparison of unbound iron-binding capacity, transferrin saturation, and C282Y genotyping in 5,211 voluntary blood donors. Hepatology 2000; 31: 1160–1164.
Allen KJ, Gurrin LC, Constantine CC et al: Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med 2008; 358: 221–230.
Altes A, Ruiz A, Barcelo MJ et al: Prevalence of the C282Y, H63D, and S65C mutations of the HFE gene in 1,146 newborns from a region of Northern Spain. Genet Test 2004; 8: 407–410.
Andrikovics H, Kalmar L, Bors A et al: Genotype screening for hereditary hemochromatosis among voluntary blood donors in Hungary. Blood Cells Mol Dis 2001; 27: 334–341.
Aranda N, Viteri FE, Fernandez-Ballart J, Murphy M, Arija V : Frequency of the hemochromatosis gene (HFE) 282C—>Y, 63H—>D, and 65S—>C mutations in a general Mediterranean population from Tarragona, Spain. Ann Hematol 2007; 86: 17–21.
Baiget M, Barcelo MJ, Gimferrer E : Frequency of the HFE C282Y and H63D mutations in distinct ethnic groups living in Spain. J Med Genet 1998; 35: 701.
Barry E, Derhammer T, Elsea SH : Prevalence of three hereditary hemochromatosis mutant alleles in the Michigan Caucasian population. Community Genet 2005; 8: 173–179.
Beutler E, Felitti V, Gelbart T, Ho N : The effect of HFE genotypes on measurements of iron overload in patients attending a health appraisal clinic.[Erratum appears in Ann Intern Med 2001;134(8):715]. Ann Intern Med 2000; 133: 329–337.
Beutler E, Felitti VJ, Koziol JA, Ho NJ, Gelbart T : Penetrance of 845G—> A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet 2002; 359: 211–218.
Burt MJ, George PM, Upton JD et al: The significance of haemochromatosis gene mutations in the general population: implications for screening. Gut 1998; 43: 830–836.
Byrnes V, Ryan E, Barrett S, Kenny P, Mayne P, Crowe J : Genetic hemochromatosis, a Celtic disease: is it now time for population screening? Genet Test 2001; 5: 127–130.
Candore G, Mantovani V, Balistreri CR et al: Frequency of the HFE gene mutations in five Italian populations. Blood Cells Mol Dis 2002; 29: 267–273.
Cimburova M, Putova I, Provaznikova H, Horak J : Hereditary hemochromatosis: detection of C282Y and H63D mutations in HFE gene by means of guthrie cards in population of Czech Republic. Genet Epidemiol 2002; 23: 260–263.
Cukjati M, Vaupotic T, Rupreht R, Curin-Serbec V : Prevalence of H63D, S65C and C282Y hereditary hemochromatosis gene mutations in Slovenian population by an improved high-throughput genotyping assay. BMC Med Genet 2007; 8: 69.
Distante S, Berg JP, Lande K, Haug E, Bell H : High prevalence of the hemochromatosis-associated Cys282Tyr HFE gene mutation in a healthy Norwegian population in the city of Oslo, and its phenotypic expression. Scand J Gastroenterol 1999; 34: 529–534.
Floreani A, Rosa RE, Basso D et al: An open population screening study for HFE gene major mutations proves the low prevalence of C282Y mutation in Central Italy. Aliment Pharmacol Ther 2007; 26: 577–586.
Guttridge MG, Carter K, Worwood M, Darke C : Population screening for hemochromatosis by PCR using sequence-specific primers. Genet Test 2000; 4: 111–114.
Hoppe C, Watson RM, Long CM et al: Prevalence of HFE mutations in California newborns. Pediatr Hematol Oncol 2006; 23: 507–516.
Jackson HA, Carter K, Darke C et al: HFE mutations, iron deficiency and overload in 10,500 blood donors. Br J Haematol 2001; 114: 474–484.
Jones DC, Young NT, Pigott C et al: Comprehensive hereditary hemochromatosis genotyping. Tissue Antigens 2002; 60: 481–488.
Kucinskas L, Juzenas S, Sventoraityte J et al: Prevalence of C282Y, H63D, and S65C mutations in hereditary HFE-hemochromatosis gene in Lithuanian population. Ann Hematol 2012; 91: 491–495.
Mariani R, Salvioni A, Corengia C et al: Prevalence of HFE mutations in upper Northern Italy: study of 1132 unrelated blood donors. Dig Liver Dis 2003; 35: 479–481.
Marshall DS, Linfert DR, Tsongalis GJ : Prevalence of the C282Y and H63D polymorphisms in a multi-ethnic control population. Int J Mol Med 1999; 4: 389–393.
Matas M, Guix P, Castro JA et al: Prevalence of HFE C282Y and H63D in Jewish populations and clinical implications of H63D homozygosity. Clin Genet 2006; 69: 155–162.
McLaren GD, Gordeuk VR : Hereditary hemochromatosis: insights from the Hemochromatosis and Iron Overload Screening (HEIRS) Study. Hematology Am Soc Hematol Educ Program 2009; 195–206.
Meier P, Schuff-Werner P, Steiner M : Hemochromatosis gene HFE Cys282Tyr mutation analysis in a cohort of Northeast German hospitalized patients supports assumption of a North to South allele frequency gradient throughout Germany. Clin Lab 2005; 51: 539–543.
Merryweather-Clarke AT, Pointon JJ, Shearman JD, Robson KJ : Global prevalence of putative haemochromatosis mutations. J Med Genet 1997; 34: 275–278.
Merryweather-Clarke AT, Simonsen H, Shearman JD, Pointon JJ, Norgaard-Pedersen B, Robson KJ : A retrospective anonymous pilot study in screening newborns for HFE mutations in Scandinavian populations. Hum Mutat 1999; 13: 154–159.
Olynyk JK, Cullen DJ, Aquilia S, Rossi E, Summerville L, Powell LW : A population-based study of the clinical expression of the hemochromatosis gene. N Engl J Med 1999; 341: 718–724.
Phatak PD, Ryan DH, Cappuccio J et al: Prevalence and penetrance of HFE mutations in 4865 unselected primary care patients. Blood Cells Mol Dis 2002; 29: 41–47.
Pozzato G, Zorat F, Nascimben F et al: Haemochromatosis gene mutations in a clustered Italian population: evidence of high prevalence in people of Celtic ancestry. Eur J Hum Genet 2001; 9: 445–451.
Andersen RV, Tybjaerg-Hansen A, Appleyard M, Birgens H, Nordestgaard BG : Hemochromatosis mutations in the general population: iron overload progression rate. Blood 2004; 103: 2914–2919.
Gurrin LC, Bertalli NA, Dalton GW et al: HFE C282Y/H63D compound heterozygotes are at low risk of hemochromatosis-related morbidity. Hepatology 2009; 50: 94–101.
Lim EM, Rossi E, De Boer WB, Reed WD, Jeffrey GP : Hepatic iron loading in patients with compound heterozygous HFE mutations. Liver Int 2004; 24: 631–636.
Ramakrishna R, Gupta S, Sarathy K, Bowen A : Phenotypic and clinical manifestations of compound heterozygous genetic haemochromatosis (CHGH): a non-invasive approach to clinical management. Intern Med J 2013; 43: 254–261.
Walsh A, Dixon JL, Ramm GA et al: The clinical relevance of compound heterozygosity for the C282Y and H63D substitutions in hemochromatosis. Clin Gastroenterol Hepatol 2006; 4: 1403–1410.
Bajaj J, Knox JF, Komorowski R, Saeian K : The irony of herbal hepatitis: Ma-Huang-induced hepatotoxicity associated with compound heterozygosity for hereditary hemochromatosis. Dig Dis Sci 2003; 48: 1925–1928.
Schoniger-Hekele M, Muller C, Polli C, Wrba F, Penner E, Ferenci P : Liver pathology in compound heterozygous patients for hemochromatosis mutations. Liver 2002; 22: 295–301.
Cheng R, Barton JC, Morrison ED et al: Differences in hepatic phenotype between hemochromatosis patients with HFE C282Y homozygosity and other HFE genotypes. J Clin Gastroenterol 2009; 43: 569–573.
Neghina AM, Anghel A : Hemochromatosis genotypes and risk of iron overload—a meta-analysis. Ann Epidemiol 2011; 21: 1–14.
Kelley M, Joshi N, Xie Y, Borgaonkar M : Iron overload is rare in patients homozygous for the H63D mutation. Can J Gastroenterol Hepatol 2014; 28: 198–202.
Aguilar-Martinez P, Bismuth M, Picot MC et al: Variable phenotypic presentation of iron overload in H63D homozygotes: are genetic modifiers the cause? Gut 2001; 48: 836–842.
Krawczak M : ASP—a simulation-based power calculator for genetic linkage studies of qualitative traits, using sib-pairs. Hum Genet 2001; 109: 675–677.
Jacobs EM, Hendriks JC, Marx JJ et al: Morbidity and mortality in first-degree relatives of C282Y homozygous probands with clinically detected haemochromatosis compared with the general population: the HEmochromatosis FAmily Study (HEFAS). Neth J Med 2007; 65: 425–433.
Powell LW, Dixon JL, Ramm GA et al: Screening for hemochromatosis in asymptomatic subjects with or without a family history. Arch Intern Med 2006; 166: 294–301.
McCune CA, Ravine D, Carter K et al: Iron loading and morbidity among relatives of HFE C282Y homozygotes identified either by population genetic testing or presenting as patients. Gut 2006; 55: 554–562.
Lazarescu A, Snively BM, Adams PC : Phenotype variation in C282Y homozygotes for the hemochromatosis gene. Clin Gastroenterol Hepatol 2005; 3: 1043–1046.
Jacobs EM HJ, van Deursen CT, Kreeftenberg HG, de Vries RA, Marx JJ, Stalenhoef AF VA, Swinkels DW : Severity of iron overload of proband determines serum ferritin levels in families with HFE-related hemochromatosis: the HEmochromatosis FAmily Study. J Hepatol 2009; 50: 174–183.
Gleeson FRE, Barrett S, Crowe J : Clinical expression of haemochromatosis in Irish C282Y homozygotes identified through family screening. Eur J Gastroenterol Hepatol 2004; 16: 859–63.
Powell LW : Diagnosis of hemochromatosis. Semin Gastrointest Dis 2002; 13: 80–88.
Elmberg M, Hultcrantz R, Simard JF, Carlsson A, Askling J : Increased risk of arthropathies and joint replacement surgery in patients with genetic hemochromatosis: a study of 3,531 patients and their 11,794 first-degree relatives. Arthritis Care Res 2013; 65: 678–685.
Nelson RL PV, Davis F, Becker E : Risk of disease in siblings of patients with hereditary hemochromatosis. Digestion 2001; 64: 120–124.
Elmberg M, Hultcrantz R, Ebrahim F et al: Increased mortality risk in patients with phenotypic hereditary hemochromatosis but not in their first-degree relatives. Gastroenterology 2009; 137: 1301–1309.
Adams P : Hemochromatosis: new insights in pathogenesis and diagnosis following the discovery of the gene. Crit Rev Clin Lab Sci 1998; 35: 239–273.
Independent UK Panel on Breast Cancer Screening: The benefits and harms of breast cancer screening:an independent review. Lancet 2012; 380: 1778–1786.
McPherson K : Screening for breast cancer - balancing the debate. BMJ 2010; 340: c3106.
Beral V, Alexander M, Duffy S et al. The number of women who would need to be screened regularly by mammography to prevent one death from breast cancer. J Med Screen 2011; 18: 210–212.
Reyes M, Dunet DO, Isenberg KB, Trisolini M, Wagener DK : Family-based detection for hereditary hemochromatosis. J Genet Couns 2008; 17: 92–100.
Aleman S, Endalib S, Stal P et al: Health check-ups and family screening allow detection of hereditary hemochromatosis with less advanced liver fibrosis and survival comparable with the general population. Scand J Gastroenterol 2011; 46: 1118–1126.
Beaton MD, Adams PC : Prognostic factors and survival in patients with hereditary hemochromatosis and cirrhosis. Can J Gastroenterol 2006; 20: 257–260.
Niederau C, Fischer R, Purschel A, Stremmel W, Haussinger D, Strohmeyer G : Long-term survival in patients with hereditary hemochromatosis. Gastroenterology 1996; 110: 1107–1119.
Bardou-Jacquet EMF, Perrin M, Manet G, Guyader D, Moirand R, Deugnier Y : Survival and Causes of Death in a cohort of 1086 treated C282Y HFE Homozygous Patients. Am J Hematol 2013; 88: E38.
Asberg A, Hveem K, Kannelonning K, Irgens WO : Penetrance of the C28Y/C282Y genotype of the HFE gene. Scand J Gastroenterol 2007; 42: 1073–1077.
Waalen J, Felitti V, Gelbart T, Ho NJ, Beutler E : Prevalence of hemochromatosis-related symptoms among individuals with mutations in the HFE gene. Mayo Clin Proc 2002; 77: 522–530.
Deugnier Y, Jouanolle AM, Chaperon J et al: Gender-specific phenotypic expression and screening strategies in C282Y-linked haemochromatosis: a study of 9396 French people. Br J Haematol 2002; 118: 1170–1178.
Adams PC, Passmore L, Chakrabarti S et al: Liver diseases in the hemochromatosis and iron overload screening study. Clin Gastroenterol Hepatol 2006; 4: 918–923, quiz 807.
Gordeuk VR, Reboussin DM, McLaren CE et al: Serum ferritin concentrations and body iron stores in a multicenter, multiethnic primary-care population. Am J Hematol 2008; 83: 618–626.
Aranda N, Viteri FE, Montserrat C, Arija V : Effects of C282Y, H63D, and S65C HFE gene mutations, diet, and life-style factors on iron status in a general Mediterranean population from Tarragona, Spain. Ann Hematol 2010; 89: 767–773.
Burke W, Imperatore G, McDonnell SM, Baron RC, Khoury MJ : Contribution of different HFE genotypes to iron overload disease: a pooled analysis. Genet Med 2000; 2: 271–277.
Waalen J, Felitti VJ, Gelbart T, Beutler E : Screening for hemochromatosis by measuring ferritin levels: a more effective approach. Blood 2008; 111: 3373–3376.
Adams PC : The natural history of untreated HFE-related hemochromatosis. Acta Haematol 2009; 122: 134–139.
Van Aken MO, De Craen AJ, Gussekloo J et al: No increase in mortality and morbidity among carriers of the C282Y mutation of the hereditary haemochromatosis gene in the oldest old: the Leiden 85-plus study. Eur J Clin Invest 2002; 32: 750–754.
Njajou OT, Houwing-Duistermaat JJ, Osborne RH et al: A population-based study of the effect of the HFE C282Y and H63D mutations on iron metabolism. Eur J Hum Genet 2003; 11: 225–231.
Rossi E, Bulsara MK, Olynyk JK, Cullen DJ, Summerville L, Powell LW : Effect of hemochromatosis genotype and lifestyle factors on iron and red cell indices in a community population. Clin Chem 2001; 47: 202–208.
Schiepers OJ, van Boxtel MP, de Groot RH et al: Serum iron parameters, HFE C282Y genotype, and cognitive performance in older adults: results from the FACIT study. J Gerontol A Biol Sci Med Sci 2010; 65: 1312–1321.
Olynyk JK, Hagan SE, Cullen DJ, Beilby J, Whittall DE : Evolution of untreated hereditary hemochromatosis in the Busselton population: a 17-year study. Mayo Clin Proc 2004; 79: 309–313.
Yamashita C, Adams PC : Natural history of the C282Y homozygote for the hemochromatosis gene (HFE) with a normal serum ferritin level. Clin Gastroenterol Hepatol 2003; 1: 388–391.
Gurrin LC, Osborne NJ, Constantine CC et al: The natural history of serum iron indices for HFE C282Y homozygosity associated with hereditary hemochromatosis. Gastroenterology 2008; 135: 1945–1952.
Allen KJ, Bertalli NA, Osborne NJ et al: HFE Cys282Tyr homozygotes with serum ferritin concentrations below 1000 microg/L are at low risk of hemochromatosis. Hepatology 2010; 52: 925–933.
Von Recklinghausen F Ueber Haemochromatose. Naturforscher Arzte Heidelberg: Tageblatt Versammlung Dische, 1989.
Sheldon J : Haemochromatosis. Oxford University Press: London: London, 1935.
Simon M, Bourel M, Genetet B, Fauchet R : Idiopathic hemochromatosis. Demonstration of recessive transmission and early detection by family HLA typing. N Engl J Med 1977; 297: 1017–1021.
Schimanski LM, Drakesmith H, Merryweather-Clarke AT et al: In vitro functional analysis of human ferroportin (FPN) and hemochromatosis-associated FPN mutations. Blood 2005; 105: 4096–4102.
Montosi G, Donovan A, Totaro A et al: Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest 2001; 108: 619–623.
Ellervik C, Birgens H, Tybjaerg-Hansen A, Nordestgaard BG : Hemochromatosis genotypes and risk of 31 disease endpoints: meta-analyses including 66,000 cases and 226,000 controls. Hepatology 2007; 46: 1071–1080.
Pedersen P, Melsen GV, Milman N : Frequencies of the haemochromatosis gene (HFE) variants C282Y, H63D and S65C in 6,020 ethnic Danish men. Ann Hematol 2008; 87: 735–740.
Pedersen P, Milman N : Extrinsic factors modifying expressivity of the HFE variant C282Y, H63D, S65C phenotypes in 1,294 Danish men. Ann Hematol 2009; 88: 957–965.
Ellard S, Lindsay H, Camm N et al. Practice guidelines for Targeted Next Generation Sequencing Analysis and Interpretation, 2012, http://www.acgs.uk.com/.
Teixeira E, Borlido-Santos J, Brissot P et al: The importance of the general practitioner as an information source for patients with hereditary haemochromatosis. Patient Educ Couns 2014; 96: 86–92.
Holmstrom P, Marmur J, Eggertsen G, Gafvels M, Stal P : Mild iron overload in patients carrying the HFE S65C gene mutation: a retrospective study in patients with suspected iron overload and healthy controls. Gut 2002; 51: 723–730.
International Organization for Standardization www.iso.org/iso/home.html.
Berwouts S, Morris MA, Dequeker E : Approaches to quality management and accreditation in a genetic testing laboratory. Eur J Hum Genet 2010; 18: S1–19.
Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS : American Association for the Study of Liver D: Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54: 328–343.
Human Genome Variation Society http://www.hgvs.org/rec.html.
Practice guidelines for the Interpretation and Reporting of Unclassified Variants (UVs) in Clinical Molecular Genetics http://cmgsweb.shared.hosting.zen.co.uk/BPGs/pdfs%20current%20bpgs/UV%20GUIDELINES%20ratified.pdf.
The Human Gene Mutation Database http://www.hgmd.cf.ac.uk/ac/index.php.
OECD Guidelines for Quality Assurance in Molecular Genetic Testing http://www.oecd.org/science/biotech/38839788.pdf.
Mutalyser https:/mutalyser.nl.
Acknowledgements
We wish to thank Prof. David Barton, Chair of the Management Board of the European Molecular Quality Network (http://www.emqn.org) for promoting the revision of the guidelines, for funding and facilitating the BPM and for funding topublish the manuscript as an open access document. We acknowledge Jacques Rochette, from the Laboratoire de Génétique, Hôpital Sud CBH, CHU-AMIENS, responsible for the French Network involved in molecular diagnosis ofhaemochromatosis, for kindly revising the first draft of this manuscript, Monique Wessels from Knowledge Institute of Medical Specialists (KiMS) in the Netherlands for her contribution in the literature search and Emerência Teixeira for her support inthe manuscript editing. We would also like to acknowledge IBMC, the Institute for Molecular and Cellular Biology, Porto, Portugal, and its staff who kindly provided the facilities and means to host there the Best Practice Meeting on the 14–15 May 2014, and thank all the participants of this meeting which resulted in the final drafting of the guidelines.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Best Practice Meeting Participants: Isabel Alonso, Alan Balfe, Rita Bastos, Nicholas Beauchamp, Fiona Blanco Kelly, Pierre Brissot, Javier Garcia-Planells, João Goncalves, Carol Hardy, Caroline Joyce, Monika Jurkowska, Steve Keeney, Lourdes Loidi, Ana Lopes, Orla Maguire, Alison May, Erica Moran Martinez, Michael Morris, Ulle Murumets, Vassos Neocleous, Graça Porto, Linda Piekuse, Serge Pissard, Noemi Roy, Jorge Sequeiros, Dorine Swinkels, Carolyn Tysoe, Martine van Belzen, Raina Yamamoto and Heinz Zoller attended the Best Practice meeting on the 14–15 May 2014 in Porto, Portugal.
Author contributions
GP led the HH guidelines project, coordinated feedback among the drafting team members, analyzed data and co-organized the Best Practice Meeting (BPM). GP, PB, DWS and HZ constituted the guidelines drafting team. They designed and validated the questionnaire on current laboratory practices used for the online survey. This was organized, analyzed and reported by OK and GP. The drafting team was responsible for the systematic literature search and/or leading the BPM discussions on the topics: the clinical value of testing for p.H63D (HZ); role, efficacy and efficiency of cascade screening (DWS); follow-up of unaffected p.C282Y homozygotes and p.C282Y/p.H63D compound heterozygotes (GP) and the value of expertise centers for HH (PB). SK revised and approved the first draft, led the BPM discussion on the topic of the future of NGS gene panels and was responsible for drafting the final version of the guidelines. SP organized the BPM. IA analyzed data, revised the manuscript and participated in the local organization of the BPM. MM actively participated in the BPM, and, in his capacity as EMQN guideline reviewer, revised and approved the final version of the document.
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Porto, G., Brissot, P., Swinkels, D. et al. EMQN best practice guidelines for the molecular genetic diagnosis of hereditary hemochromatosis (HH). Eur J Hum Genet 24, 479–495 (2016). https://doi.org/10.1038/ejhg.2015.128
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.128
This article is cited by
-
Diagnostic and prognostic relevance of using large gene panels in the genetic testing of patients with dilated cardiomyopathy
European Journal of Human Genetics (2023)
-
Carrier burden of over 300 diseases in Han Chinese identified by expanded carrier testing of 300 couples using assisted reproductive technology
Journal of Assisted Reproduction and Genetics (2023)
-
Multiparametric MR mapping in clinical decision-making for diffuse liver disease
Abdominal Radiology (2020)
-
Erythroferrone, the new iron regulator: evaluation of its levels in Egyptian patients with beta thalassemia
Annals of Hematology (2020)
-
Quantification of liver iron overload disease with laser ablation inductively coupled plasma mass spectrometry
BMC Medical Imaging (2018)
