
Abstract
Rett syndrome (RTT), a neurodevelopmental disorder that predominantly affects females, is primarily caused by variants in MECP2. Variants in other genes such as CDKL5 and FOXG1 are usually associated with individuals who manifest distinct phenotypes that may overlap with RTT. Individuals with phenotypes suggestive of RTT are typically screened for variants in MECP2 and then subsequently the other genes dependent on the specific phenotype. Even with this screening strategy, there are individuals in whom no causative variant can be identified, suggesting that there are other novel genes that contribute to the RTT phenotype. Here we report a de novo deletion of protein tyrosine phosphatase, non-receptor type 4 (PTPN4) in identical twins with a RTT-like phenotype. We also demonstrate the reduced expression of Ptpn4 in a Mecp2 null mouse model of RTT, as well as the activation of the PTPN4 promoter by MeCP2. Our findings suggest that PTPN4 should be considered for addition to the growing list of genes that warrant screening in individuals with a RTT-like phenotype.
Similar content being viewed by others


Introduction
Although variants in MECP2 account for the majority of Rett syndrome (RTT) cases, diagnosis is still based on clinical criteria.1, 2 Typical RTT is characterised by a period of developmental regression usually commencing in infancy, which is followed by a recovery or stabilisation phase. Affected individuals exhibit impairment or complete loss of language and hand skills, have gait abnormalities, and display stereotypic hand movements. The current clinical criteria are outlined in Neul et al2 and take into account the variable nature of the RTT phenotype and allow a better description of the variant forms of RTT that are typically difficult to define clinically.
Despite intensive investigations of both RTT patients and Mecp2 mutant mouse models, the biological functions of MeCP2 and mechanisms of disease are still not fully understood. What has become apparent is that MeCP2 has multifaceted roles that influence normal brain development and function.3, 4 Initially, MeCP2 was thought to be a transcriptional regulator of a subset of genes key to the modulation of synaptic plasticity; however, defining these genes has proven somewhat problematic.3, 4, 5, 6, 7 Although it is apparent that MeCP2 binds to the promoters of a number of genes such as BDNF 8 and JUNB,9 it is also postulated that MeCP2 may also act in a more global fashion by modulating chromatin architecture.10
In addition to variants in MECP2, other molecular causes of RTT and related phenotypes have been identified, including CDKL5,11, 12, 13 FOXG114 and TCF4,15 although currently there is growing support to re-categorise individuals with CDKL5 and FOXG1 variants as being distinct clinical entities,16, 17 and TCF4 variants generally cause a discrete phenotype, Pitt–Hopkins syndrome.18 These genes may share biological pathways with MECP2, thereby potentially underpinning the overlapping phenotype. In this study, we identify a novel deletion of protein tyrosine phosphatase, non-receptor type 4 (PTPN4) in twins with an atypical form of RTT.
Materials and methods
CGH array
Analysis of copy number variation (CNV) of the twins and their parents was carried out by Sydney Genome Diagnostics, the Children’s Hospital at Westmead, using an Agilent Sureprint G3Hmn CGH 400K array, 60mer oligo probes, effective resolution 0.08 Mb (Agilent Technologies, Mulgrave, VIC, Australia). The identity panel PowerPlex 16 (Promega, Alexandria, NSW, Australia) was also used to confirm that the twins were monozygous.
Quantitation of Ptpn4 expression
Brains were harvested from Mecp2tm1Tam mice (Mecp2+/y and Mecp2−/y) at 5 weeks (presymptomatic) and 8–10 weeks (symptomatic), and the hippocampus and cerebellum was dissected from each brain. RNA extraction, cDNA synthesis, and quantitative PCR (qPCR) were performed as described previously;19 for primers, see Supplementary Methods. The expression levels of Ptpn4 were calculated relative to Gapdh using the ΔΔCT method.20
Chromatin immunoprecipitation
Differentiated SH-SY5Y cells were used for chromatin immunoprecipitation (ChIP) assays. Chromatin fragments were prepared by sonication and used to perform MeCP2 immunoprecipitation (Supplementary Methods). The ability of MeCP2 to bind to target DNA regions was then measured by qPCR; 1 μl of eluted DNA (as 5 μl of a 1:5 dilution) was added, used as a template, and reactions were performed in triplicate. For primers, see Supplementary Methods. Fold enrichment was calculated as fold enrichment relative to the input sample, and the ΔCT was calculated using the average CT of the INPUT sample as the calibrating sample (sample CT—CT of the INPUT sample).
Luciferase assay
The human PTPN4 promoter (g.119758859-119759770), NC_00002.12, was PCR amplified using Platinum Pfx DNA Polymerase (Life Technologies, Mulgrave, VIC, Australia). The PCR product was cloned into the Luciferase reporter vector pcBG99-Basic vector (Promega) using HindIII producing the pCBG99-PTPN4 luciferase reporter construct, and the sequence and orientation of the PTPN4 promoter was confirmed by sequencing. SH-SY5Y cells were plated into six-well plates (2 × 106 cells per well) and transfected the following day using Lipofectamine LTX (Life Technologies). The ability of MeCP2 to act as a transcriptional regulator of the PTPN4 promoter was tested by transfecting 4 μg of the pcBG99-PTPN4 or pcBG99-basic and 4 μg of a MeCP2 expression vector (pCMV-Tag2C-MeCP2) or the empty expression vector (pCMV-Tag2) simultaneously into the SH-SY5Y cells. In addition, all cells were transfected with 2 μg of the red luciferase vector pcBR99, as a transfection control. Twenty-four hours after transfection, the cells were harvested and lysed with Glo Lysis buffer (Promega). Equal volumes (50 μl) of the lysate and the luciferase substrate, Chroma-Glo Luciferase Assay System (Promega), were combined in a 96-well white/black plate, and the luminescence of the red and green luciferases was measured immediately using the VICTOR3 plate reader (Perkin Elmer, Melbourne, VIC, Australia). The red luciferase was measured with the filter 610AGLP while the green was measured with the filter 510DF60, and the raw red and green luminescence values were corrected as recommended by the manufacturer (see Supplementary Methods). For each sample, the relative amount of green luminescence was calculated by dividing the green luminescence by the red luminescence. Two independent experiments with triplicate transfections and duplicate readings were performed. In order to compare the results of the two independent experiments, the relative green luminescence of the pcBG99-PTPN4 vector in the presence of MeCP2 was divided by the average of the relative green luminescence of the pcBG99-PTPN4 vector without MeCP2.
Statistical analyses
qPCR data of Ptpn4 transcripts and ChIP enrichment and relative luminescence values of the Luciferase reporter system were analysed for statistical significance using the Mann–Whitney test and Graph Pad Prism, version 5.03 (GraphPad Software, San Diego, CA, USA).
Database submission
The novel PPNT4 deletion identified in this study was submitted to the DECIPHER database, with the accession numbers being 294047 for Twin 1 and 294048 for Twin 2; https://decipher.sanger.ac.uk/.
Results
Case study
Identical twins (confirmed using the PowerPlex 16 identity panel) were born at 36 weeks gestation after a normal pregnancy and delivery. Twin A’s birth weight was 2670 gm (50th percentile), length was 44.5 cm (10th–50th percentiles) and head circumference was 33.5 cm (50th percentile). Twin B’s birth weight was 2575 gm (10th–50th percentile), length was 44.5 cm (10th–50th percentile) and head circumference was 33 cm (10th–50th percentile). There were no perinatal or postnatal problems, but they were described as being quiet and placid babies. Both were poor feeders and slow to gain weight. First concerns about developmental progress were raised at 9 months, as neither twin had rolled or started to crawl, and they were described as being floppy.
In general, Twin A’s development has always been ahead of Twin B. She sat without support at 12 months, walked unaided at 3 years and 4 months, developed a pincer grip at 2 years and started to say single words between 5 and 6 years of age. She has never lost any speech, could talk in short sentences and could imitate words; however, speech was often difficult to comprehend. She had some stereotypic hand movements, putting her hands to her head, previously to her mouth, flexing and extending her fingers and rubbing and twisting her fingers. There was no loss of fine motor skills. At the age of 17 years, Twin A could throw, catch and kick a ball.
Twin B sat unsupported at 15 months, walked unaided at 4 years and 4 months, developed at pincer grip at 2 years and started saying single words at 6 years. She lost the ability to speak from approximately 9 years of age. She had learnt to say approximately six words but, at age 17 years, did not articulate any words. She had good receptive language skills and could follow simple commands.
Both twins had a seizure disorder from approximately 2½ years of age. Twin A’s EEG at the time showed symmetric background 8–10 Hz activity posteriorly. Epileptiform activity was present with frequent sharp/spike discharges noted in the left occipital region. An EEG performed at 10 years showed poorly formed background activity and mutlti-focal sharp discharges bilaterally. There were frequent spike and wave generalised paroxysmal discharges, most without clinical accompaniments. She was seizure free from 13 years of age. Anticonvulsant medications were subsequently stopped.
Twin B’s EEG performed at 12 years showed frequent polyspike discharges, which alternated from the right to left hemisphere. At 13 years, video telemetry documented partial seizures with an onset from the left frontotemporal region. The interictal EEG showed continuous generalised slowing of the background activity, which was more prominent on the left. At 17 years, Twin B continued to have generalised seizures, some associated with cyanosis lasting up to 2 min every 6–8 weeks. This was managed with sodium valproate and lamotrogine.
The twins had ECGs performed at 17 years of age. Twin A had a prolonged corrected QT value but no cardiac arrhythmia seen on 24-h Holter monitoring, while twin B’s ECG was normal.
Both twins had a normal breathing pattern. They had a mildly disturbed but relatively good sleep pattern, with occasional walking at night, laughing, talking and singing. They used to grind their teeth, but this subsequently spontaneously ceased. They had some difficulty chewing, in that they chewed with an open mouth, and Twin B rolled food around with her tongue. They were described as fussy eaters but did not require their food to be softened, pureed or mashed. Both had constipation.
When reviewed at age 17 years, Twin A appeared in good health. She was alert and cooperative throughout the consultation and at times spoke in sentences. Her weight was 36.5 kg (10th percentile), height 140.3 cm (<1st percentile) and head circumference was 53 cm (~10th percentile). She had mild trigonocephaly, a high nasal bridge, almond shaped eyes, prominent lips, thickened gums, widely spaced teeth and a narrow palate. She had shortened 4th metatarsals. Her feet were cool to palpation and mottled. Her back was straight and hirsute. She could walk without assistance with a slightly wide-based gait. She repetitively twisted an object in her hand. She drooled but did not grind her teeth.
When last reviewed with her sister, Twin B also appeared in good health. Her weight was 26.1 kg (<3rd percentile), height was 132.1 cm (<1st percentile) and head circumference was 51 cm (2nd percentile). Like her sister, she had mild trigonocephaly, a high wide nasal bridge, almond shaped eyes, thickened gums and widely spaced teeth. Her back was straight and hirsute. She had small cold hands and feet with shortened 4th metatarsals bilaterally. She demonstrated stereotypic hand movements in the form of putting her hands to the side of her head, flexing and extending her fingers continuously and flapping her hands. She was able to walk unaided with a wide base of support.
Overall, although the twins have many of the features suggestive of RTT they did not fulfil the clinical criteria for the typical form of the disorder. Twin B could be classified as having variant RTT according to the Neul criteria;2 however, Twin A did not fulfil the clinical criteria for variant RTT.
De novo deletion at chromosome 2q14.2
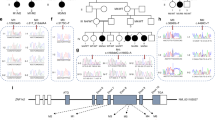
CGH array analysis of the twins identified a deletion (arr[hg19] 2q14.2(120,584,760-120,726,563) × 1 dn), with a minimum size of 90 Kb (Chr2.hg19:g. 120,584,760_120,726,563del) and a maximum size of 160 Kb (Chr2.hg19:g. 120,579,294 _120,734,950del), Figure 1. This deletion was not seen in their healthy parents or their two healthy siblings. This region contains a single gene, protein tyrosine phosphatase, non-receptor type 4 (PTPN4/PTPMEG), GenBank ref NC_000002. This phosphatase is expressed predominately in the brain and testis21 and knockout of the Drosophila and murine orthologues results in neurological abnormalities.22, 23 This essential role of PTPN4 in neuronal development and function suggests that PTPN4 is a good candidate gene for the variant RTT phenotype seen in the twins.

Comparative genomic hybridisation (CGH) array analysis. CGH array testing mapped the approximate position of the deletion, at coordinates 120.58–120.70 Mb, within chromosome 2. The 400K array data shown here is for Twin A. The central grey bar indicates gene PTPN4. The x axis shows the hg19 coordinates. This image is derived from the CGH Fusion software: InfoQuant Ltd (London, UK). Note that the segmentation algorithm used here may have underestimated the distal extent of the deletion (see arrow). Within the ISCN terminology, the deletion was reported as extending from 120,584,760 to 120,726,563.
No benign CNVs involving PTPN4 have been identified in the Copy Number Variation Healthy Control Database (http://cnv.chop.edu). Searches of the CNV database DECIPHER24 identified 10 individuals with deletions or duplications involving the PTPN4 (Table 1). Two of the individuals with de novo deletions displayed intellectual disability, and one with a de novo deletion was described as having oculomotor apraxia, although there are little additional clinical details for the latter case. However, all of these cases had deletions encompassing multiple genes, and therefore their phenotypes cannot necessarily be attributed to PTPN4 alone.
Ptpn4 is downregulated in the brain of a mouse model of Rett syndrome
Several Mecp2 knockout and knock-in mice have been developed to model the RTT phenotype. Although the phenotype of the Ptpn4 null mouse is mild compared with that of the Mecp2 null mouse, both models display impairment of motor co-ordination and learning. We therefore examined whether Ptpn4 expression is dysregulated in the brain of the Mecp2tm1Tam mouse. qPCR analysis of the Ptpn4 transcript (Figure 2) demonstrated a significant reduction of Ptpn4 levels in both cerebellum and hippocampus of symptomatic Mecp2−/y mice, while in the presymptomatic animals a significant reduction was only observed in the cerebellum but not in the hippocampus. A brain region-specific dysregulation of Ptpn4 in presymptomatic Mecp2−/y mice thus became more widespread with progression of the disease phenotype.

Ptpn4 expression reduced in the brain of the Mecp2tm1Tam mouse model of RTT. Quantitation of Ptpn4 expression in the cerebellum and hippocampus of presymptomatic and symptomatic Mecp2y/+ (wild type) and Mecp2y/− (null) mice (n=5, error bars=SD).
MeCP2 binds to PTPN4 sequences
MeCP2 is a transcriptional regulator that can both activate and repress gene expression. As Ptpn4 expression is reduced in the absence of Mecp2, it is possible that MeCP2 may function to activate the expression of PTPN4. To test whether PTPN4 is a transcriptional target, we tested for MeCP2 binding to PTPN4 sequences using ChIP assays in the SH-SY5Y human neuroblastoma cell line. Two PCR amplicons were designed to amplify regions 5′ to exon 1 of PTPN4, containing the putative RNA polymerase II binding sites and the CpG island, respectively. An additional amplicon in intron 15 of PTPN4 was also designed as a gene-specific non-promoter sequence control (Figure 3a). As a positive control for MeCP2 binding, an amplicon in the promoter of a known MeCP2 target in SH-SH5Y cells, JUNB,9 was tested. For a negative control, intron 6 of PORIN was used. The MeCP2 ChIP studies revealed enrichment for all amplicons above that of the normal serum and no antibody control background levels (Figure 3b).

ChIP analysis of MeCP2-binding sites in SH-SY5Y cells. (a) Location of the amplicons in the PTPN4 gene. (b) Fold enrichment of MeCP2, no antibody and normal mouse serum ChIP from SH-SY5Y lysates across the PTPN4, JUNB and PORIN amplicons. Duplicate biological replicates were analysed in triplicate, P-values for each amplicon relative to the no antibody control (NS=not significant, **P≤0.01, ***P≤0.001). Error bars=SD.
MeCP2 enhances the expression of the PTPN4 promoter in vitro
Given the inconclusive results of the ChIP assays, we next investigated the ability of MeCP2 to mediate the expression PTPN4 promoter using a luciferase reporter vector containing the PTPN4 promoter (pcBG99-PTPN4). In SH-SY5Y cells, the level of luciferase expression in the presence of MeCP2 was 2.1±0.67-fold of that of pcBG99-PTPN4 alone (Figure 4), indicating that MeCP2 is capable of acting as a transcriptional activator to the PTPN4 promoter.

MeCP2 enhances PTPN4 promoter-driven gene expression in SHSY-5Y cells. Relative luminescence of the promoterless luciferase vector (pcBG99-basic) and the PTPN4 promoter-driven luciferase vector (pcBG99-PTPN4), with and without MeCP2. Two independent experiments with triplicate transfections and duplicate readings were performed. Error bars=SD.
Discussion
This is the first report of a human disease associated with haplo-insufficiency of PTPN4. Evidence from knockout models in other species demonstrates that PTPN4 is required for neuronal development and function. In Drosophila, the null phenotype manifests as a defect of axonal development,23 while knockout mice show impaired motor learning and cerebellar synaptic plasticity.22 Although essential for neuronal function, the phenotype of the Ptpn4 knockout mouse is milder than that of Mecp2 null mice.22, 25, 26, 27
Similarly, compared with typical RTT, the twins described here have a milder phenotype. It is interesting to note that while the twins exhibited a number of clinical features that overlap with those seen in RTT, including shortening of the 4th metatarsals, only the more severely affected twin fulfilled the clinical criteria for variant RTT. A potentially important distinguishing feature is the facial dysmorphism exhibited by the twins, which is not seen in MECP2 mutation-positive RTT individuals, but it remains to be seen whether these dysmorphic features are a consequence of PTPN4 haplo-insufficiency.
PTPN4 is predominantly expressed in the brain, with the strongest expression in the thalamus and the Purkinje cells of the cerebellum.21 PTPN4 is a phosphatase that regulates the phosphorylation of the glutamate receptor GluRє1/GRIN2A in Purkinje neurons and is capable of interacting with both GRIN2A and another glutamate receptor GluRdelta2/GRID2.21 Variations in GRIN2A are associated with autism spectrum disorders, sporadic schizophrenia28, 29 and epileptic encephalopathy30while GRID2 is important for regulating synaptic plasticity and motor co-ordination.31 The phenotype seen in the Ptpn4 knockout mouse could be due to a modulation of the function of these glutamate receptors.
Although deficiency of MeCP2 function is the major cause of classical RTT, the exact pathological mechanisms have been elusive.3, 4 However, there are clearly defects of dendritic spine number and structure in the Mecp2-deficient brain, which result in a generalised loss of synaptic plasticity.32, 33, 34, 35 In comparison, Ptpn4 null mice display a more specific loss of cerebellar synaptic plasticity,22 consistent with the more restricted expression of Ptpn4. The observed reduction of Ptnp4 in the brain of Mecp2 null mice and the in vitro demonstration that MeCP2 enhances the strength of the PTPN4 promoter in neuronal cells suggests this phosphatase may be a component of the biological pathways that are disrupted by Mecp2 deficiency.
In conclusion, we describe a de novo deletion of the gene PTPN4 in identical twins with a RTT-like phenotype. Furthermore, we demonstrate reduced expression of Ptpn4 in the brain of a Mecp2 null mouse model and the activation of PTPN4 promoter by MeCP2. Together, these results suggest that haplo-insufficiency of PTPN4 may be the cause of the phenotype seen in these twins. We suggest that PTPN4 should be considered as a possible novel candidate gene of variant RTT.
References
Hagberg B : Clinical delineation of Rett syndrome variants. Neuropediatrics 1995; 26: 62.
Neul JL, Kaufmann WE, Glaze DG et al: Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 2010; 68: 944–950.
Guy J, Cheval H, Selfridge J, Bird A : The role of MeCP2 in the brain. Annu Rev Cell Dev Biol 2011; 27: 631–652.
Samaco RC, Neul JL : Complexities of Rett syndrome and MeCP2. J Neurosci 2011; 31: 7951–7959.
Nuber UA, Kriaucionis S, Roloff TC et al: Up-regulation of glucocorticoid-regulated genes in a mouse model of Rett syndrome. Hum Mol Genet 2005; 14: 2247–2256.
Traynor J, Agarwal P, Lazzeroni L, Francke U : Gene expression patterns vary in clonal cell cultures from Rett syndrome females with eight different MECP2 mutations. BMC Med Genet 2002; 3: 12.
Tudor M, Akbarian S, Chen RZ, Jaenisch R : Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci USA 2002; 99: 15536–15541.
Martinowich K, Hattori D, Wu H et al: DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003; 302: 890–893.
Yasui DH, Peddada S, Bieda MC et al: Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci USA 2007; 104: 19416–19421.
Skene PJ, Illingworth RS, Webb S et al: Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell 2010; 37: 457–468.
Bahi-Buisson N, Nectoux J, Rosas-Vargas H et al: Key clinical features to identify girls with CDKL5 mutations. Brain 2008; 131: 2647–2661.
Mari F, Azimonti S, Bertani I et al: CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet 2005; 14: 1935–1946.
Weaving LS, Christodoulou J, Williamson SL et al: Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet 2004; 75: 1079–1093.
Roche-Martinez A, Gerotina E, Armstrong-Moron J, Sans-Capdevila O, Pineda M : [FOXG1, a new gene responsible for the congenital form of Rett syndrome]. Rev Neurol 2011; 52: 597–602.
Armani R, Archer H, Clarke A et al: Transcription factor 4 and myocyte enhancer factor 2C mutations are not common causes of Rett syndrome. Am J Med Genet A 2012; 158A: 713–719.
Fehr S, Wilson M, Downs J et al: The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur J Hum Genet 2012; 21 (3): 266–273.
Kortum F, Das S, Flindt M et al: The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis. J Med Genet 2011; 48: 396–406.
Zweier C, Peippo MM, Hoyer J et al: Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome). Am J Hum Genet 2007; 80: 994–1001.
Williamson SL, Giudici L, Kilstrup-Nielsen C et al: A novel transcript of cyclin-dependent kinase-like 5 (CDKL5) has an alternative C-terminus and is the predominant transcript in brain. Human Genet 2012; 131: 187–200.
Schmittgen TD, Livak KJ : Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008; 3: 1101–1108.
Hironaka K, Umemori H, Tezuka T, Mishina M, Yamamoto T : The protein-tyrosine phosphatase PTPMEG interacts with glutamate receptor delta 2 and epsilon subunits. J Biol Chem 2000; 275: 16167–16173.
Kina S, Tezuka T, Kusakawa S et al: Involvement of protein-tyrosine phosphatase PTPMEG in motor learning and cerebellar long-term depression. Eur J Neurosci 2007; 26: 2269–2278.
Whited JL, Robichaux MB, Yang JC, Garrity PA : Ptpmeg is required for the proper establishment and maintenance of axon projections in the central brain of Drosophila. Development 2007; 134: 43–53.
Firth HV, Richards SM, Bevan AP et al: DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 2009; 84: 524–533.
Chen RZ, Akbarian S, Tudor M, Jaenisch R : Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet 2001; 27: 327–331.
Guy J, Hendrich B, Holmes M, Martin JE, Bird A : A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet 2001; 27: 322–326.
Pelka GJ, Watson CM, Radziewic T et al: Mecp2 deficiency is associated with learning and cognitive deficits and altered gene activity in the hippocampal region of mice. Brain 2006; 129: 887–898.
Tarabeux J, Kebir O, Gauthier J et al: Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl Psychiatry 2011; 1: e55.
Yoo HJ, Cho IH, Park M, Yang SY, Kim SA : Family based association of GRIN2A and GRIN2B with Korean autism spectrum disorders. Neurosci Lett 2012; 512: 89–93.
Endele S, Rosenberger G, Geider K et al: Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet 2010; 42: 1021–1026.
Kakegawa W, Miyoshi Y, Hamase K et al: D-serine regulates cerebellar LTD and motor coordination through the delta2 glutamate receptor. Nat Neurosci 2011; 14: 603–611.
Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM : Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis 2006; 21: 217–227.
Lonetti G, Angelucci A, Morando L et al: Early environmental enrichment moderates the behavioral and synaptic phenotype of Mecp2 null mice. Biol Psychiatry 2010; 67: 657–665.
Moretti P, Levenson JM, Battaglia F et al: Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci 2006; 26: 319–327.
Zhang L, He J, Jugloff DG, Eubanks JH : The Mecp2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus 2008; 18: 294–309.
Acknowledgements
This work was supported by the National Health and Medical Research Council (NHMRC) of Australia (Project Grant 457238), the Rett Syndrome Association of New South Wales, the Rett Syndrome Australian Research Fund and the Rett Syndrome Association of Australia. GJP was an NHMRC Biomedical (Peter Doherty) Fellow. PPLT is a NHMRC Senior Principal Research Fellow (Grant 1003100).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Williamson, S., Ellaway, C., Peters, G. et al. Deletion of protein tyrosine phosphatase, non-receptor type 4 (PTPN4) in twins with a Rett syndrome-like phenotype. Eur J Hum Genet 23, 1171–1175 (2015). https://doi.org/10.1038/ejhg.2014.249
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.249
