
Abstract
Dentinogenesis imperfecta is an autosomal dominant disease characterized by severe hypomineralization of dentin and altered dentin structure. Dentin extra cellular matrix is composed of 90% of collagen type I and 10% of non-collagenous proteins among which dentin sialoprotein (DSP), dentin glycoprotein (DGP) and dentin phosphoprotein (DPP) are crucial in dentinogenesis. These proteins are encoded by a single gene: dentin sialophosphoprotein (DSPP) and undergo several post-translational modifications such as glycosylation and phosphorylation to contribute and to control mineralization. Human mutations of this DSPP gene are responsible for three isolated dentinal diseases classified by Shield in 1973: type II and III dentinogenesis imperfecta and type II dentin dysplasia. Shield classification was based on clinical phenotypes observed in patient. Genetics results show now that these three diseases are a severity variation of the same pathology. So this review aims to revise and to propose a new classification of the isolated forms of DI to simplify diagnosis for practitioners.
Similar content being viewed by others
Introduction
Today, molecular biology and genetics studies increase our knowledge about dental germ formation and give a wider view of spatio-temporal patterns of gene expressions and roles. Mutations of these genes explained some of the dental diseases only clinically described before. Structural diseases of mineral tissue may have a primary genetic or a secondary acquired aetiology, with local or general origin. Structural damages diffuse (the whole tooth is affected), extend (all the teeth are affected) and define a hereditary aetiology. A family history of the pathology is often observed, however, sporadic cases may occur.1
Dentin genetic diseases are known for several years and many reviews have been published.2, 3 They include two entities: dentinogenesis imperfecta (DGI) and dentin dysplasia (DD).2, 4 The only epidemiological data available was published in 1975. According to Witkop,5 dentinogenesis imperfecta estimated incidence was between 1/6000 and 1/8000 and dentin dysplasia 1/100 000. These data only came from United States. In other countries, case reports concerned smaller cohorts. Clinical classification used at that time was also old and may be outdated in view of recent information (Shield et al6) (Table 1). Shield classified dentinogenesis imperfecta into three subgroups: one associated with a syndrome osteogenesis imperfecta (DGI type I) and two isolated (DGI type II and III); he also described two dentin dysplasia (DD type I and II). In the literature, some case reports described clinical signs that belonged to several of these diseases, so diagnosis and classification were very difficult for practitioners. Therefore, clinical manifestations were insufficient to determine if two pathologies or a differential expression of the same pathology are present. With identification of the key gene of dentinal structure, the dentin sialophosphoprotein (DSPP), authors reported that Shield classification has become obsolete but never proposed a new one.7 This study proposes to establish a more accurate classification that separates syndromic and isolated forms taking into account the variability of expression because of gene variants.8
Dentinogenesis and dental biomineralization
Dentin is a mesenchymal cellular mineralized tissue. Odontoblasts are fully differentiated cells and do not proliferate. The cell bodies are located at the pulp periphery with their odontoblast processes running through dentin to reach enamel dentin junction.9, 10
Dentin formation consists of odontoblast secretion of an extracellular matrix, the predentin, followed by mineralization. This matrix is composed of 90% of type I collagen and 10% of non-collagenous proteins (NCP) and lipids. NCP regroup: proteins, phosphoproteins, proteoglycans, growths factors, enamel proteins, phosphatases (alkaline phosphatase), proteases (matrix metalloproteinases and their inhibitors: MMPs and TIMPs), that interact and build the scaffold that will support and initiate mineralization.11
DSPP encodes for a single mRNA giving three proteins cleaved in specific sites11, 12 by proteases from astacin and MMP families:9, 13 DSP (dentin sialoprotein), DGP (dentin glycoprotein) and DPP (dentin phosphoprotein).14, 15, 16, 17 DSPP-related peptides are the major components of non-collagenous proteins and believed to have a crucial role in converting predentin into mineralized dentin. DSP is a sialic acid-rich proteoglycan with few phosphorylations; conversely, DPP is highly phosphorylated and exhibits numerous isoforms depending on their phosphorylation degree.16, 18 DPP binds to collagen fibrils in specific area (between the gap and overlap zone) of hydroxyapatite nucleation.19, 20 In vitro, immobilized on a stable support and incubated in a solution containing relevant concentration of calcium and phosphate, DPP initiates hydroxyapatite formation at low concentration and inhibits growth at higher concentration.21 This process adapts mineralization to specific conditions for initiating, height and length growth of hydroxyapatite crystals.21, 22 DPPcKO mice analysis revealed that DPP rescues predentin formation compared with Dspp null mice.23 On the other hand, DSP has still unclear functions but is involved in mineral nucleation.17, 24, 25, 26
The DSPPs are encoded by a 5-exon gene. Exons 2, 3, 4 and the beginning of exon 5 encode for DSP. DGP and DPP are encoded by the end of exon 5. DSPP expression is restricted to differentiated odontoblasts (with transient expression in presecretory ameloblast).14, 27 So, DSPP-related peptides were initially qualified as dentinal-specific proteins. However, later, low levels of DSPP transcripts have been detected in mouse inner ear,28 periodontal ligament,29 and bone ( × 400 less).30, 31
Isolated hereditary dentinal diseases
Dentinogenesis imperfecta (Shield Type II)
The most frequent dentinal disorder is characterized by an autosomal dominant inheritance. The teeth exhibit a grey blue, or amber brown and opalescent discolouration and small height. Enamel is often dislodged because of altered enamel dentin junction. Consequently, the exposed hypomineralized dentin is fast worn out by attrition. Attrition ranges from few facets of erosion to a complete disappearance of the crown. This influences dental surgery, undertaken frequently on realizing an overdenture in temporary dentition.32 Primary teeth are often more severely affected than permanent teeth (Figure 1). The radiographical aspect is pathognomonic: bulbous crowns because of an important cervical constriction, short and thick roots and pulp obliteration. Study of the reported cases shows a widest variability: either total or partial obliteration of pulp chamber and/or root whatever dentition concerned.33, 34 Periodontal diseases are frequent without carious disease. Proposed aetiologies are enamel and dentinal permeability for pathological bacteria or necrosis induced by pulpal obliteration. Nevertheless, the interpretation was not satisfactory because it did not take into account the cellular and molecular features of this pathology. Indeed, Silva et al35 proved that dentin proteins induced neutrophil recruitment under pathology. Histological studies of these teeth showed few and large dentinal tubules running in various directions with atubular area. Enamel fractures occurred along stries of Retzius where prisms are discutinued.36 Enamel dentin junction is also altered, this explains the phenomenon of enamel chipping. High-resolution synchrotron radiation computed tomography and small angle X-ray scattering showed anomalies of shape and thickness of crystallites.37

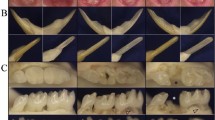
Clinical phenotype of moderate dentinogenesis imperfecta (formerly DGI Shield type II) in temporary teeth of 6-year-old boy (a–c) and permanent teeth at 8 years old (d–f), primary teeth show brown opalescent discoloration, severe attrition, complete pulp obliteration of erupted teeth, bulbous crown, short root, periapical radiolucency without carie. Temporary teeth are more severely affected than permanent teeth.
A candidate gene for DGI-II was identified on chromosome 4q21 by linkage analysis between markers D4S2691 and D4S2692.38 Xiao et al28 described the first mutation in association with the disease. The causing gene encodes a structural protein of the dentin, the DSPP, which is involved in mineralization process. This gene belongs to a genes cluster called SIBLING’S (Small Integrin Binding Ligant N-Linked glycoprotein) implicated in mineralization of dental and bone tissues notably by modulation of matrix metalloproteinases activity.39
Dentinogenesis imperfecta (Type III de Shield)
This pathology is very rare, quite exclusively described in the triracial sub-population of Maryland, the ‘Brandywine isolate’. This population has the highest incidence of all dental genetic diseases at 1 in 15.40 Clinically, it is like DGI-II. It affects the two dentitions, with blue amber opalescent teeth, bulbous crowns, and important attrition. But the major difference comes from the pulpal aspect. They were qualified as ‘shell teeth’ because of an important pulp enlargement on dental X-rays. Pulp is apparent through occlusal dentin and exposures are frequent. The pulpal enlargement results in a reduction in dentin tissue.41 However, DGI-II and DGI-III could be found on subjects of the same family, and even in the same patient.42 Dong et al43 showed a DSPP variant in a Brandiwyne DGI-III family confirming the allelic relation between DGI-II and DGI-III.
Dentin dysplasia type I or radicular
This dentinal defect is extremely rare (1/100 000)5 and concerns the two dentitions. Clinically, teeth appear normal. The first sign of disease is tooth mobility that leads to premature exfoliations of teeth either spontaneously or with minor traumatisms.44 Radiographical aspect, classified by O’Carroll et al,45 is characteristic: roots are shorter (absent in severe cases), merged (taurodont) with an apical conical aspect, with sharp construction (Figure 2). Likewise, premature teeth lost with root length alterations signs this pathology and allow to discriminate DD-I and hypophosphatasia. Pulp is replaced by a mineralized tissue with a dentin aspect: completely or partially with crescent-shaped horizontal radiolucent lines. Periapical lesions are frequent without any other associated pathology. The shorter the root, the more the pulp is obliterated and the greater the prevalence of periodontal pathology.

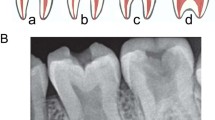
Clinical phenotype of radicular dentinal dysplasia (formerly DD Shield type I): teeth show normal colour (a) and shape but roots are severely shortened with conical apex and molar are taurodont (b) (arrow).
These teeth were described without colour alterations. However, a careful examination of the literature reveals that this criterion is not characteristic. Some authors describe discoloration: brown/blue, yellow/grey,46, 47 or just a brown opacity at the incisal level.47, 48
This pathology is transmitted as an autosomal dominant trait with complete penetrance.49 Until now the causing gene is unknown and the physiopathological process not understood. Several hypotheses have been mentioned: an alteration in the epithelial part of tooth germ during root morphogenesis inducing a premature invagination of the Hertwig root sheat and a premature stop.50, 51 Nevertheless, this hypothesis of epithelial alteration is not accepted unanimously. Others implicate the mesenchymal part of teeth, perhaps the problem lies in odontoblast differentiation or functions at the root level.
Dentin dysplasia type II or coronary
DD-II is inherited as an autosomal dominant trait. Clinically, this pathology has the same expression as Shield type II dentinogenesis imperfecta but affects only deciduous teeth. Permanent teeth are normal in colour, shape and height.52 However, on X-rays, they exhibit a pulp aspect in ‘thistle-tube’, meaning a large pulpal chamber prolonged by thin root canals (Figure 3). In addition, pulpal stones are frequent.53, 54 DD-II is, therefore, similar to DGI-II and they can be differentiated only by their clinical appearance in permanent teeth. No data concerning DD-II prevalence is reported in the literature, and only few case reports are published. So, we could conclude that this phenotype is very rare. A careful examination of these cases shows that DGI-II is often present in other members of the family related to the patient or in the patient himself with also discoloration of their permanent teeth.55 An allelic relation between these two pathologies was proposed by McDougall38 and Dean based on linkage analysis.56 Description of DSPP mutation in a patient affected by DD-II confirmed this hypothesis and explained these clinical similarities.57

Clinical phenotype of mild dentinogenesis imperfecta (formerly DD Shield type II) in temporary teeth of a 4-year-old girl (a–c) and permanent teeth at 10 years old (d–f), primary teeth show amber opalescent discoloration, attrition of first molar, partial to complete pulp obliteration of erupted teeth, bulbous crown and short root. Permanent teeth show normal colour without attrition. (g) Panoramic radiograph of their mother showing typical ‘thistle pulp’ aspect, constriction at enamel cement junction (arrow), short root and periapical radiolucency without carie (*).
Towards a nosology defined by mutations spectrum
To date 38 pathological variants have been described in the DSPP gene, located in the peptide signal region, within the DSP region and within the DPP region (Table 2).1, 7, 28, 43, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75 Most of them (28) leads to DGI-II, eight to DD-II and two to DGI-III. No mutation has been described within the DGP coding region. According to many authors, these three pathologies correspond to an expression spectrum of the same physiopathological entity.2, 7, 76 Nevertheless, none of them proposed an update of the Shield classification.
To propose a new classification, we analysed the clinical spectrum of DSPP variants related to the disease based on Witkop and Shield diagnosis criteria (Table 2). The analysis of all the reported cases with DSPP variants showed variable expression of the clinical and radiographical features. The crown discoloration was ranged from slight grey, blue-grey, amber to brown. Crowns showed a short and bulbous aspect. Different degrees of attrition ranging from enamel chipping to whole crown disappearance were noted. Finally, roots appeared thinner and shorter. Pulp chamber and root canal obliteration varied from enlargement, partial obliteration with ‘thistle-shaped’ aspect, narrowed pulp or complete obliteration. Periapical radiolucency may be present or absent. All these diagnosis criteria define a continuum of the phenotypic features from mild, moderate to severe expression (Table 3). Most of the reported cases showed a moderate disease with blue grey amber opalescent teeth, bulbous and short crown, attrition, thinner and shorter roots, and partial or total pulp obliteration. Periapical radiolucency was not constant within the reports. The increased severity of the clinical features in primary teeth when compared with the permanent teeth, usually described in DD-II, DGI-II and DGI-III, was confirmed in all the reviewed cases. Primary teeth showed mainly moderate and severe alterations whereas permanent teeth showed mild to severe alterations. Mild forms were described solely in permanent teeth and defined by the absence or a slight discoloration of teeth, few attrition, and partial pulp obliteration with ‘thistle-shaped’ aspect. These cases were previously diagnosed as DD-II. DD-II is known to be the less severe phenotype which validated our classification.
Dspp null mice, diagnosed as DGI-III, could be seen as the most severe phenotype of the DSPP disease.77 Indeed, the mice are characterized by crown discolorations, increased predentin, dentin hypomineralization, severe attrition and shortened teeth roots. Pulp enlargement giving shell teeth aspect was the pathognomonic feature of this phenotype. Enlargement of pulp was described in five cases in primary teeth, and in one patient in permanent teeth. Enlarged pulp in primary teeth in very young children (2–4 years old in all the five DGI-III cases) has to be carefully considered since it is known that obliteration might progress and permanent denture has to be waited for definitive diagnosis. In permanent teeth, severe phenotype was described in a Brandywine patient diagnosed for DGI-III confirming the proposed classification of severe DI (Table 2).43 In this case, DNA variant affects the serine repeat and aspartic acid-rich region, especially important to calcium and phosphate interactions that are essential for mineralization. The expression variability, even in the same family, is a constant observation in this hereditary condition. The same DSPP variant could result in a moderate or severe phenotype, previously diagnosed for DG-II or/and DGI-III. Correlating genotype/phenotype is not an easy task if we consider the possible interference of environmental factors. Moreover, in DSPP disease only one gene but three proteins are in cause. Differential functions and levels of expression (DSP to DPP molar ratio 1:10)78 of each protein complicate the interpretation of the findings.
The present analysis revealed that DSPP-related disease showed variable expression severity of the same entity. We propose that all DSPP diseases are called exclusively ‘Dentinogenesis Imperfecta (DI)’ characterized by grey to brown crowns, shortened and bulbous crowns, shorter and thinner roots, attrition and pulp alterations. The severity can be ranged from mild to severe forms. Mild forms are characterized by no or very few signs in permanent teeth except ‘thisthle like’ aspect of pulp and a moderate affection of primary teeth. Severe forms are characterized by the presence of all DI features with pulp enlargement especially in permanent teeth.
This present analysis simplifies dentinogenesis imperfecta diagnosis taking account the clinical variability and may really help practitioners. Shield type II dentin dysplasia may be considered as a mild form and Shield type III dentinogenesis imperfecta as a severe form of DI (Table 1). (Of course, Shield DGI-I, which is syndromic, also called osteogenesis imperfecta is not concerned by this new classification since it is a whole different pathology). Dentin dysplasia Shield type I becomes the unique radicular pathology named radicular dentin dysplasia. Its aetiology is still unknown.
Conclusion
Dentin gene diseases are since long described in the literature from the clinical viewpoint. Like many hereditary pathologies, they are very rare, but according to Witkop,40 they occur more often than enamel diseases. A large revision about all of these epidemiological data would give us new data about this observation that concerned only US population, in 1970. Still now, isolated dentinal genetical diseases regroup four entities among which three look like each other clinically. Molecular genetics studies allow to discriminate only two pathologies: dentinogenesis imperfecta (formerly DGI Shield type II, III and DD Shield type II) and dentin dysplasia (formerly DD Shield type I) that correspond to a radicular anomaly. After syndromic exclusion (no bone alteration especially), isolated dentinogenesis imperfecta should actually be regarded as a single entity with an important variability of expression following our new classification. This disease is caused by the DSPP gene variants, encoding the major non-collagenous proteins, which are implicated in initiation and growth of hydroxyapatite crystals. Radicular dentin dysplasia aetiology and physiopathology are still obscure.
All variants published and presented in Table 2 can be consulted on the LOVD website (http://www.lovd.nl/DSPP).
Abbreviations
DSPP, Dentin sialophosphoprotein; DSP, Dentin sialoprotein; DPP, Dentin phosphoprotein; DGP, Dentin glycoprotein; DGI/DI, dentinogenesis imperfecta; DD, dentin dysplasia; MM, Pmatrix metaloproteinase; NCP, non-collagenous protein; TIMP, tissue inhibitor metalloproteinase; SIBLING’S, Small Integrin Binding Ligant N-Linked glycoprotein.
References
Zhang J, Wang J, Ma Y et al: A novel splicing mutation alters DSPP transcription and leads to dentinogenesis imperfecta type II. PLoS One 2011; 6: e27982.
Hart PS, Hart TC : Disorders of human dentin. Cells Tissues Organs 2007; 186: 70–77.
Kim JW, Simmer JP : Hereditary dentin defects. J Dent Res 2007; 86: 392–399.
Barron MJ, McDonnell ST, Mackie I, Dixon MJ : Hereditary dentine disorders: dentinogenesis imperfecta and dentine dysplasia. Orphanet J Rare Dis 2008; 3: 31.
Witkop Jr CJ : Hereditary defects of dentin. Dent Clin North Am 1975; 19: 25–45.
Shields ED, Bixler D, el-Kafrawy AM : A proposed classification for heritable human dentine defects with a description of a new entity. Arch Oral Biol 1973; 18: 543–553.
McKnight DA, Simmer JP, Hart PS, Hart TC, Fisher LW : Overlapping DSPP mutations cause dentin dysplasia and dentinogenesis imperfecta. J Dent Res 2008; 87: 1108–1111.
MacDougall M, Dong J, Acevedo AC : Molecular basis of human dentin diseases. Am J Med Genet 2006; 140: 2536–2546.
Linde A, Goldberg M : Dentinogenesis. Crit Rev Oral Biol Med 1993; 4: 679–728.
Ruch JV, Lesot H, Begue-Kirn C : Odontoblast differentiation. Int J Dev Biol 1995; 39: 51–68.
Butler WT, Brunn JC, Qin C, McKee MD : Extracellular matrix proteins and the dynamics of dentin formation. Connect Tissue Res 2002; 43: 301–307.
MacDougall M, Simmons D, Luan X, Nydegger J, Feng J, Gu TT : Dentin phosphoprotein and dentin sialoprotein are cleavage products expressed from a single transcript coded by a gene on human chromosome 4. Dentin phosphoprotein DNA sequence determination. J Biol Chem 1997; 272: 835–842.
Yamakoshi Y, Hu JC, Iwata T, Kobayashi K, Fukae M, Simmer JP : Dentin sialophosphoprotein is processed by MMP-2 and MMP-20 in vitro and in vivo. J Biol Chem 2006; 281: 38235–38243.
Begue-Kirn C, Ruch JV, Ridall AL, Butler WT : Comparative analysis of mouse DSP and DPP expression in odontoblasts, preameloblasts, and experimentally induced odontoblast-like cells. Eur J Oral Sci 1998; 106 (Suppl 1): 254–259.
Feng JQ, Luan X, Wallace J et al: Genomic organization, chromosomal mapping, and promoter analysis of the mouse dentin sialophosphoprotein (Dspp) gene, which codes for both dentin sialoprotein and dentin phosphoprotein. J Biol Chem 1998; 273: 9457–9464.
Ritchie HH, Wang L : The presence of multiple rat DSP-PP transcripts. Biochim Biophys Acta 2000; 1493: 27–32.
Liu Q, Gibson MP, Sun H, Qin C : Dentin sialophosphoprotein (DSPP) plays an essential role in the postnatal development and maintenance of mouse mandibular condylar cartilage. J Histochem Cytochem 2013; 61: 749–758.
Ritchie HH, Li X : A novel rat dentin mRNA coding only for dentin sialoprotein. Eur J Oral Sci 2001; 109: 342–347.
Beniash E, Traub W, Veis A, Weiner S : A transmission electron microscope study using vitrified ice sections of predentin: structural changes in the dentin collagenous matrix prior to mineralization. J Struct Biol 2000; 132: 212–225.
Huq NL, Loganathan A, Cross KJ et al: Association of bovine dentine phosphophoryn with collagen fragments. Arch Oral Biol 2005; 50: 807–819.
Boskey AL, Maresca M, Doty S, Sabsay B, Veis A : Concentration-dependent effects of dentin phosphophoryn in the regulation of in vitro hydroxyapatite formation and growth. Bone Miner 1990; 11: 55–65.
Saito T, Arsenault AL, Yamauchi M, Kuboki Y, Crenshaw MA : Mineral induction by immobilized phosphoproteins. Bone 1997; 21: 305–311.
Suzuki S, Sreenath T, Haruyama N et al: Dentin sialoprotein and dentin phosphoprotein have distinct roles in dentin mineralization. Matrix Biol 2009; 28: 221–229.
Boskey A, Spevak L, Tan M, Doty SB, Butler WT : Dentin sialoprotein (DSP) has limited effects on in vitro apatite formation and growth. Calcif Tissue Int 2000; 67: 472–478.
Liu Y, Han D, Wang L, Feng H : Down-regulation of Wnt10a affects odontogenesis and proliferation in mesenchymal cells. Biochem Biophys Res Commun 2013; 434: 717–721.
Yamakoshi Y, Hu JC, Fukae M et al: Porcine dentin sialoprotein is a proteoglycan with glycosaminoglycan chains containing chondroitin 6-sulfate. J Biol Chem 2005; 280: 1552–1560.
Ritchie HH, Berry JE, Somerman MJ et al: Dentin sialoprotein (DSP) transcripts: developmentally-sustained expression in odontoblasts and transient expression in pre-ameloblasts. Eur J Oral Sci 1997; 105: 405–413.
Xiao S, Yu C, Chou X et al: Dentinogenesis imperfecta 1 with or without progressive hearing loss is associated with distinct mutations in DSPP. Nat Genet 2001; 27: 201–204.
Baba O, Qin C, Brunn JC et al: Detection of dentin sialoprotein in rat periodontium. Eur J Oral Sci 2004; 112: 163–170.
Butler WT, Brunn JC, Qin C : Dentin extracellular matrix (ECM) proteins: comparison to bone ECM and contribution to dynamics of dentinogenesis. Connect Tissue Res 2003; 44 (Suppl 1): 171–178.
Qin C, Brunn JC, Cadena E, Ridall A, Butler WT : Dentin sialoprotein in bone and dentin sialophosphoprotein gene expressed by osteoblasts. Connect Tissue Res 2003; 44 (Suppl 1): 179–183.
Walter JD : The use of overdentures in patients with dentinogenesis imperfecta. J Paediatr Dent 1988; 4: 17–25.
Malmgren B, Lundberg M, Lindskog S : Dentinogenesis imperfecta in a six-generation family. A clinical, radiographic and histologic comparison of two branches through three generations. Swed Dent J 1988; 12: 73–84.
Acevedo AC, Santos LJ, Paula LM, Dong J, MacDougall M : Phenotype characterization and DSPP mutational analysis of three Brazilian dentinogenesis imperfecta type II families. Cells Tissues Organs 2009; 189: 230–236.
Silva TA, Lara VS, Silva JS, Oliveira SH, Butler WT, Cunha FQ : Macrophages and mast cells control the neutrophil migration induced by dentin proteins. J Dent Res 2005; 84: 79–83.
Wright JT, Gantt DG : The ultrastructure of the dental tissues in dentinogenesis imperfecta in man. Arch Oral Biol 1985; 30: 201–206.
Kinney JH, Pople JA, Driessen CH, Breunig TM, Marshall GW, Marshall SJ : Intrafibrillar mineral may be absent in dentinogenesis imperfecta type II (DI-II). J Dent Res 2001; 80: 1555–1559.
MacDougall M : Refined mapping of the human dentin sialophosphoprotein (DSPP) gene within the critical dentinogenesis imperfecta type II and dentin dysplasia type II loci. Eur J Oral Sci 1998; 106 (Suppl 1): 227–233.
Fisher LW, Fedarko NS : Six genes expressed in bones and teeth encode the current members of the SIBLING family of proteins. Connect Tissue Res 2003; 44 (Suppl 1): 33–40.
Witkop CJ : Hereditary defects in enamel and dentin. Acta Genet Stat Med 1957; 7: 236–239.
Sapir S, Shapira J : Dentinogenesis imperfecta: an early treatment strategy. Pediatr Dent 2001; 23: 232–237.
Heimler A, Sciubba J, Lieber E, Kamen S : An unusual presentation of opalescent dentin and Brandywine isolate hereditary opalescent dentin in an Ashkenazic Jewish family. Oral Surg Oral Med Oral Pathol 1985; 59: 608–615.
Dong J, Gu T, Jeffords L, MacDougall M : Dentin phosphoprotein compound mutation in dentin sialophosphoprotein causes dentinogenesis imperfecta type III. Am J Med Genet 2005; 132A: 305–309.
Comer TL, Gound TG : Hereditary pattern for dentinal dysplasia type Id: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2002; 94: 51–53.
O’Carroll MK, Duncan WK, Perkins TM : Dentin dysplasia: review of the literature and a proposed subclassification based on radiographic findings. Oral Surg Oral Med Oral Pathol 1991; 72: 119–125.
Ansari G, Reid JS : Dentinal dysplasia type I: review of the literature and report of a family. ASDC J Dent Child 1997; 64: 429–434.
Steidler NE, Radden BG, Reade PC : Dentinal dysplasia: a clinicopathological study of eight cases and review of the literature. Br J Oral Maxillofac Surg 1984; 22: 274–286.
Kalk WW, Batenburg RH, Vissink A : Dentin dysplasia type I: five cases within one family. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1998; 86: 175–178.
O Carroll MK, Duncan WK : Dentin dysplasia type I. Radiologic and genetic perspectives in a six-generation family. Oral Surg Oral Med Oral Pathol 1994; 78: 375–381.
Duncan WK, Perkins TM, O Carroll MK, Hill WJ : Type I dentin dysplasia: report of two cases. Ann Dent 1991; 50: 18–21.
Melnick M, Levin LS, Brady J : Dentin dysplasia type I: a scanning electron microscopic analysis of the primary dentition. Oral Surg Oral Med Oral Pathol 1980; 50: 335–340.
Brenneise CV, Conway KR : Dentin dysplasia, type II: report of 2 new families and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1999; 87: 752–755.
Burkes Jr EJ, Aquilino SA, Bost ME : Dentin dysplasia II. J Endod 1979; 5: 277–281.
Jasmin JR, Clergeau-Guerithault S : A scanning electron microscopic study of dentin dysplasia type II in primary dentition. Oral Surg Oral Med Oral Pathol 1984; 58: 57–63.
Ranta H, Lukinmaa PL, Knif J : Dentin dysplasia type II: absence of type III collagen in dentin. J Oral Pathol Med 1990; 19: 160–165.
Dean JA, Hartsfield Jr JK, Wright JT, Hart TC : Dentin dysplasia, type II linkage to chromosome 4q. J Craniofac Genet Dev Biol 1997; 17: 172–177.
Rajpar MH, Koch MJ, Davies RM, Mellody KT, Kielty CM, Dixon MJ : Mutation of the signal peptide region of the bicistronic gene DSPP affects translocation to the endoplasmic reticulum and results in defective dentine biomineralization. Hum Mol Genet 2002; 11: 2559–2565.
Bai H, Agula H, Wu Q et al: A novel DSPP mutation causes dentinogenesis imperfecta type II in a large Mongolian family. BMC Med Genet 2010; 11: 23.
Holappa H, Nieminen P, Tolva L, Lukinmaa PL, Alaluusua S : Splicing site mutations in dentin sialophosphoprotein causing dentinogenesis imperfecta type II. Eur J Oral Sci 2006; 114: 381–384.
Kida M, Tsutsumi T, Shindoh M, Ikeda H, Ariga T : De novo mutation in the DSPP gene associated with dentinogenesis imperfecta type II in a Japanese family. Eur J Oral Sci 2009; 117: 691–694.
Kim JW, Hu JC, Lee JI et al: Mutational hot spot in the DSPP gene causing dentinogenesis imperfecta type II. Hum Genet 2005; 116: 186–191.
Lee KE, Kang HY, Lee SK et al: Novel dentin phosphoprotein frameshift mutations in dentinogenesis imperfecta type II. Clin Genet 2011; 79: 378–384.
Lee SK, Hu JC, Lee KE, Simmer JP, Kim JW : A dentin sialophosphoprotein mutation that partially disrupts a splice acceptor site causes type II dentin dysplasia. J Endod 2008; 34: 1470–1473.
Lee SK, Lee KE, Hwang YH et al: Identification of the DSPP mutation in a new kindred and phenotype-genotype correlation. Oral Dis 2011; 17: 314–319.
Lee SK, Lee KE, Jeon D et al: A novel mutation in the DSPP gene associated with dentinogenesis imperfecta type II. J Dent Res 2009; 88: 51–55.
Lee SK, Lee KE, Song SJ, Hyun HK, Lee SH, Kim JW : A DSPP mutation causing dentinogenesis imperfecta and characterization of the mutational effect. BioMed Res Int 2013; 2013: 948181.
Malmgren B, Lindskog S, Elgadi A, Norgren S : Clinical, histopathologic, and genetic investigation in two large families with dentinogenesis imperfecta type II. Hum Genet 2004; 114: 491–498.
McKnight DA, Suzanne Hart P, Hart TC et al: A comprehensive analysis of normal variation and disease-causing mutations in the human DSPP gene. Hum Mutat 2008; 29: 1392–1404.
Nieminen P, Papagiannoulis-Lascarides L, Waltimo-Siren J et al: Frameshift mutations in dentin phosphoprotein and dependence of dentin disease phenotype on mutation location. J Bone Miner Res 2011; 26: 873–880.
Song Y, Wang C, Peng B et al: Phenotypes and genotypes in 2 DGI families with different DSPP mutations. Oral Surg Oral Med Oral Pathol Oral RadiolEndod 2006; 102: 360–374.
Zhang X, Chen L, Liu J et al: A novel DSPP mutation is associated with type II dentinogenesis imperfecta in a Chinese family. BMC Med Genet 2007; 8: 52.
Zhang X, Zhao J, Li C et al: DSPP mutation in dentinogenesis imperfecta Shields type II. Nat Genet 2001; 27: 151–152.
Song YL, Wang CN, Fan MW, Su B, Bian Z : Dentin phosphoprotein frameshift mutations in hereditary dentin disorders and their variation patterns in normal human population. J Med Genet 2008; 45: 457–464.
Kim JW, Nam SH, Jang KT et al: A novel splice acceptor mutation in the DSPP gene causing dentinogenesis imperfecta type II. Hum Genet 2004; 115: 248–254.
Wang H, Hou Y, Cui Y et al: A novel splice site mutation in the dentin sialophosphoprotein gene in a Chinese family with dentinogenesis imperfecta type II. Mutat Res 2009; 662: 22–27.
Maciejewska I, Chomik E : Hereditary dentine diseases resulting from mutations in DSPP gene. J Dent 2012; 40: 542–548.
Sreenath T, Thyagarajan T, Hall B et al: Dentin sialophosphoprotein knockout mouse teeth display widened predentin zone and develop defective dentin mineralization similar to human dentinogenesis imperfecta type III. J Biol Chem 2003; 278: 24874–24880.
Ritchie HH, Ritchie DG, Wang LH : Six decades of dentinogenesis research. Historical and prospective views on phosphophoryn and dentin sialoprotein. Eur J Oral Sci 1998; 106 (Suppl 1): 211–220.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
de La Dure-Molla, M., Philippe Fournier, B. & Berdal, A. Isolated dentinogenesis imperfecta and dentin dysplasia: revision of the classification. Eur J Hum Genet 23, 445–451 (2015). https://doi.org/10.1038/ejhg.2014.159
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.159
This article is cited by
-
Establishment of a clinical network for children with amelogenesis imperfecta and dentinogenesis imperfecta in the UK: 4-year experience
European Archives of Paediatric Dentistry (2024)
-
Identification of DSPP novel variants and phenotype analysis in dentinogenesis dysplasia Shields type II patients
Clinical Oral Investigations (2023)
-
Mouse Dspp frameshift model of human dentinogenesis imperfecta
Scientific Reports (2021)
-
VPS4B mutation impairs the osteogenic differentiation of dental follicle cells derived from a patient with dentin dysplasia type I
International Journal of Oral Science (2020)
-
Dentinogenesis imperfecta in Osteogenesis imperfecta type XI in South Africa: a genotype–phenotype correlation
BDJ Open (2019)
