
Abstract
Immunodeficiency with centromeric instability and facial anomalies (ICF) syndrome is a primary immunodeficiency, predominantly characterized by agammaglobulinemia or hypoimmunoglobulinemia, centromere instability and facial anomalies. Mutations in two genes have been discovered to cause ICF syndrome: DNMT3B and ZBTB24. To characterize the clinical features of this syndrome, as well as genotype–phenotype correlations, we compared clinical and genetic data of 44 ICF patients. Of them, 23 had mutations in DNMT3B (ICF1), 13 patients had mutations in ZBTB24 (ICF2), whereas for 8 patients, the gene defect has not yet been identified (ICFX). While at first sight these patients share the same immunological, morphological and epigenetic hallmarks of the disease, systematic evaluation of all reported informative cases shows that: (1) the humoral immunodeficiency is generally more pronounced in ICF1 patients, (2) B- and T-cell compartments are both involved in ICF1 and ICF2, (3) ICF2 patients have a significantly higher incidence of intellectual disability and (4) congenital malformations can be observed in some ICF1 and ICF2 cases. It is expected that these observations on prevalence and clinical presentation will facilitate mutation-screening strategies and help in diagnostic counseling.
Similar content being viewed by others


Introduction
Immunodeficiency with centromeric instability and facial anomalies (ICF syndrome) (MIM no. 242860) is a rare autosomal recessive disease, characterized by immunodeficiency of variable extent, mild facial anomalies and chromosome instability, involving the pericentromeric regions of chromosomes 1, 9 and 16. Facial dysmorphisms may include a round face, flat nasal bridge, hypertelorism, epicanthus, up-turned nose, macroglossia, micrognathia and low-set ears. The majority of ICF patients have a delay in walking and speech development. The intelligence status is variable.1
Most patients suffer from hypogammaglobulinemia, or agammaglobulinemia, which is the immunological hallmark of ICF syndrome. Circulating B-cells in ICF patients have been reported to contain an increased proportion of immature cells, a lack of memory cells and are more prone to undergo apoptosis upon in vitro activation. Interestingly, activation, differentiation and immunoglobulin class-switch recombination driven by stimulation via the B-cell receptor and CD40 appeared to be normal.2 Studies on T-cell function are limited, and reported data suggest a normal proliferative response upon mitogenic stimulation, the capability to support Pokeweed mitogen (PWM)-induced immunoglobulin production by control B-cells and a somewhat increased degree of apoptosis.3, 4 Therefore, the relative contribution of an intrinsic B-cell defect and a defective T-cell function to the frequently observed dysgammaglobulinemia in patients with ICF syndrome remains to be elucidated.
Approximately 50% of the ICF cases carry mutations in the DNA methyltransferase 3B gene (DNMT3B) at chromosome 20q11.2.5, 6 These cases have been designated as ICF1 patients.1 No genotype–phenotype correlation was observed among patients with and without DNMT3B mutations in an earlier study.1 Recently, mutations in the zinc-finger and BTB domain-containing 24 gene (ZBTB24) on chromosome 6q21 were described in most DNMT3B mutation-negative patients, and these cases were designated as ICF2 patients.7, 8 Mutations in DNMT3B or ZBTB24 do not explain all ICF patients and there remains a small group with unknown etiology,7 here provisionally designated as ICFX. In ICF patients, large, often centromeric, DNA repeats show reduced CpG methylation, and ICF2 and ICFX patients differ from ICF1 patients by the presence of additional α-satellite repeat hypomethylation.7, 9
The aim of the present study is to identify possible differences in the clinical presentation and immunological characteristics of patients with ICF1, ICF2 and ICFX syndrome.
Materials and methods
Patients
At present, 66 patients have been included in the ICF registry. A computer-assisted literature search using PubMed and EM-base was conducted in order to identify and obtain data on patients with ICF syndrome. Supplementary information was obtained from questionnaires completed by their referring physicians. In 22 patients, molecular analysis was not performed; they were excluded from this study. Data of patients previously reported in the literature were updated (n=37) and new patients (n=7) have been included. Ethical approval was obtained for the publication of these data.
Data collected included age at diagnosis, facial anomalies, psychomotor development, hypotonia and gastrointestinal problems. The diagnosis of immunodeficiency was primarily based on reductions of serum IgG, IgG subclasses, IgA and/or IgM levels compared to age-matched controls.10 Agammaglobulinemia was defined as a decrease of IgG level below 2.5 g/l. Special attention was drawn to frequency and type of infections, and laboratory results reflecting the immunological status. Apart from serum immunoglobulin levels, complete blood count, numbers of B-cell and T-cell subpopulations at diagnosis and latest follow-up were included as well. Normal ranges for lymphocyte numbers and subpopulations are from published data.11
Mutation analysis of DNMT3B and ZBTB24
For all new patients, all coding exons and intron–exon junctions for DNMT3B and ZBTB24 were amplified from gDNA isolated from peripheral blood by PCR and the PCR products were subjected to Sanger sequencing (LGTC, Leiden, the Netherlands) as previously described.7 All described variants are based on the reference DNMT3B (NM_006892.3) and ZBTB24 (NM_014797.2) accessions.
In vitro expansion of T-cells
Peripheral blood mononuclear cells (PBMCs) from patients and parents or controls were isolated using a Ficoll-Isopaque gradient. To generate T-cell lines, 5 × 105 or 1 × 106 PBMCs were stimulated by polyclonal activation with 1 μg/ml phytohaemagglutinin (PHA; Welcome Diagnostics, Dartford, UK), and irradiated allogeneic PBMCs (3000 rad) in RPMI 1640 (Life Technologies Europe, Bleiswijk, the Netherlands) supplemented with 10% human AB serum, 20 IU/ml recombinant interleukin-2 (rIL-2; Novartis International, Basel, Switzerland), 100 IU/ml streptomycin, 100 IU/ml penicillin and 2 mM L-glutamine. The number of viable T-cells were counted in a Bürker counting chamber at day 7 or 9 following stimulation. An aliquot of 1 × 106 of the responding T-cells was re-stimulated with PHA, rIL-2 and irradiated allogeneic PBMCs. After 7 or 9 days of re-stimulation, the number of responding T-cells was determined again. Cell expansion was defined as the number of cells present at the end of the 7–9-days culture period related to the number of cells at the start of the first or second stimulation, respectively, which was set at 1.
Results
A total of 44 patients were included in the study: 23 with mutations in DNMT3B (ICF1), 13 with mutations in ZBTB24 (ICF2) and 8 without detectable mutations in either gene (ICFX) (Table 1). ICF1 patients 15 and 16, 29 and 33, 35 and 36, 51 and 52, ICF2 patients 37 and 38, 62 and 63 and 64, and ICFX patients 13 and 14, 34 and 53 are siblings. Sociodemographic and genetic data are summarized in Table 1. Dysmorphic features, developmental and neurological complications of the disease, infectious diseases and occurrence of malignancies for the patients in each group are given in Table 2.
Genetics
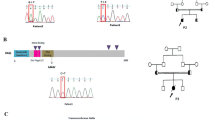
A new DNMT3B (homozygous) mutation c.1918G>C (p.G640R) was identified in patient 50 and we identified an already described homozygous mutation, c.2450A>G (p.D817G), in patient 47. In ZBTB24, homozygous mutations c.759C>G (p.T253X) and c.958C>T (p.R320X) were found in patients 40 and 55, respectively, of which the mutation found in patient 40 has not been described before. Most ICF1 patients with mutations in DNMT3B carry missense mutations in or near the catalytic domain (Table 1; Figure 1). None are homozygous for nonsense alleles. In contrast, the majority of ICF2 patients have homozygous mutations in ZBTB24. There does not seem to be a mutational hotspot in ZBTB24 and most mutations are predicted to create a premature stop codon (Table 1; Figure 1).

Schematic representation of the DNMT3B and ZBTB24 proteins, and their domains with the mutations identified in ICF1 and ICF2 patients included in this study.
Facial anomalies
Facial anomalies were observed in nearly all patients within the three groups and the pattern of facial anomalies was overlapping between ICF1, ICF2 and ICFX (Table 2). Only patient 25 in group 1 had no facial anomalies, even when he grew older. Hypertelorism, flat nasal bridge and epicanthus were the most common anomalies in all three groups.
Growth and development
Failure to thrive occurred in some patients within all groups. Macronodular cirrhosis developed in ICF1 patient 42 following treatment for acute lymphoblastic leukemia and granulomatous hepatitis in ICF2 patient 54. Motor delay was observed in ∼50% of ICF1 patients, but in nearly every ICF2 patient. Speech delay was observed in most patients of all the three groups. In addition, intellectual disability was found in about half of the ICF1patients (9/20), but in all patients with ICF2 (13/13) (P=0.001; χ2 test). Several patients with ICFX were also intellectually disabled.
Congenital malformations
Congenital malformations were reported in seven patients with ICF1. Cardiac anomalies were reported in three patients (two with a ventricular septal defect and one with atrium septal defect). Cleft lip, clinodactyly and syndactyly, choanal stenosis, hip dislocation and a horseshoe kidney were all mentioned once in a patient. In ICF2, cardiac anomalies were reported as well: once an atrium septal defect and once an ascending aorta dilatation. Congenital hypothyroidism affected ICF1 patient 42.
Cerebral malformations
Cerebral malformations, including corpus callosum hypoplasia and macrocephaly, were reported in several ICF1 patients, and cortical atrophy was mentioned in four patients. Focal cortical heterotopy has been reported in ICF2 patient 40 as well as in ICFX patient 41.12 ICF2 patient 65 had a large cerebral arachnoidal cyst.13 ICF1 patient 42 had a rod/cone retinal dystrophy. It is unclear whether this was coincidental.
Infections
Severe infections (pneumonia, sepsis) occurred in majority of the patients of all three groups. Opportunistic infections (Candida albicans, Pneumocystis jiroveci) were found in some patients within all groups (Table 2).
Malignancy
Angiosarcoma has been described in one ICF1 patient14 and ICF1 patient 42 suffered from an acute lymphoblastic leukemia. Hodgkin lymphoma was described in ICF2 patient 37.1
Immunodeficiency
Hypogammaglobulinemia or agammaglobulinemia was observed in all but one ICF1 patients (Table 3, Supplementary Figure 1). This boy, patient 2, had normal serum IgG and IgM levels.15 Agammaglobulinemia occurred in at least 14 patients. All 20 ICF1 patients with known data were IgA deficient. Three had normal IgM levels.
In ICF2, one patient had normal serum IgA and IgG levels, concomitant with a normal distribution of IgG subclasses (patient 65, Table 3).13 Of twelve ICF2 patients, half had hypogammaglobulinemia, whereas the other half had agammaglobulinemia. IgA was present in (sub)normal levels in seven patients. Likewise in seven patients, IgM was present in a (sub)normal level, whereas four patients had both IgA and IgM at normal concentrations (Table 3).
Six ICFX patients showed agammaglobulinemia (Table 3). One girl (patient 13) with ICFX had only IgM deficiency, but some years later she developed agammaglobulinemia during encephalitis due to JC virus. Her older brother (patient 14) had normal serum immunoglobulins.
In young children, the numbers of CD3+, CD4+ and CD8+ cells were normal both in ICF1 and ICF2 groups (Supplementary Table 1 and Supplementary Figure 2). T-cells become deficient in older children and young adults with ICF1. Four of the ICF1 patients developed a neutropenia and thrombocytopenia (patients 16, 25, 30 and 45) in the second decade; B- and T-cells were decreased as well (Supplementary Figure 2). Bone marrow of patient 16 showed hypoplasia.
Cell expansion after stimulation of PBMC’s containing comparable numbers of T-cells with PHA/IL-2 and irradiated allogeneic feeders for 7 or 9 days was significantly reduced in those ICF patients investigated (ICF1: patients 42 and 45, ICF2: patients 49, 54, 55 and 65) compared with unrelated controls, parents or an unaffected sibling (P=0.028; Wilcoxon test, Table 4). Significantly reduced expansion was also observed in a second round of stimulation for 7 or 9 days when ICF1 and ICF2 were considered as one group (Table 4).
Treatment
Hematopoietic stem cell transplantation (HSCT) was performed in three unrelated ICF1 patients (patients 24, 33 and 43) and in two siblings (patients 34 and 53) with ICFX. Patient 24 received a HSCT because of myelodysplasia; she died from a RSV infection. In the other patients, HSCT was performed due to severe infections associated with the immunodeficient status and was successful in all cases.16 Remarkably, HSCT was not performed in ICF2 patients.
Discussion
The hallmarks of ICF syndrome are the triad of immunodeficiency, centromeric instability and facial dysmorphisms. Not surprisingly, these are present in all three groups. Specifically, patients with ICF1 or ICF2 were found to have a similar phenotype. They have the same facial anomalies, a frequent occurrence of developmental delay and a high incidence of severe respiratory and opportunistic infections. These symptoms are also present in ICFX patients with unresolved gene defect. However, despite these common characteristics, subtle differences in immune defects, congenital malformation and intellectual function were observed among the three groups.
Immunodeficiency is severe in ICF syndrome and most patients die at young age, usually in the first or second decade. However, humoral immunodeficiency is generally more pronounced in patients with ICF1 compared with ICF2. In ICF1, all but one patients suffer from agammaglobulinemia or hypogammaglobulinemia, and all are IgA deficient. An ICF1 case with a mild phenotype was recently also reported to have a homozygous c.2308A>G (pK770E) mutation in DNMT3B.8 In ICF2, immunoglobulin class deficiencies are less extreme: one patient has normal serum immunoglobulins and six of them have normal levels of IgA and/or IgM. Blanco-Betancourt et al.2 studied two ICF1 patients and two DNMT3B mutation-negative ICF patients; three of these patients had agammaglobulinemia. The peripheral blood of these patients contained only naive B-cells, with an immature phenotype, possibly due to an accumulation of new bone marrow B-cell emigrants but no memory B-cells. They proposed that disturbance of peripheral B-cell maturation contributes to agammaglobulinemia in ICF syndrome. In vitro, B-cells of the patients were competent in class-switch recombination and immunoglobulin secretion upon stimulation via CD40L in the presence of IL-4. In line with these data, IgA and IgG are present in some ICF2 patients in this study. In one patient (patient 65), the switch is (nearly) normal, based on normal serum IgA and IgG levels (Table 3). Notably, the latter patient is the only ICF2 case reported to date harboring two missense mutations in ZBTB24: all other ICF2 patients have at least one premature stop codon. This indicates the crucial role of ZBTB24 in antibody production.
With respect to cellular immunity, the number of T-cells is normal in young ICF1 and ICF2 children (below the age of 10 years). However, once older with a long follow-up period, ICF1 patients (patients 16, 25, 30 and 45) display a decline in the numbers of T-cells in the second decade of life, when they develop neutropenia and thrombocytopenia as well (Supplementary Figure 2). Interestingly, it has been suggested that these features, together with development of cerebral atrophy, are reminiscent of systemic lupus erythematosus (SLE),1 which is linked to DNA hypomethylation.17 However, mRNA levels of DNMT3B in T-cells of SLE patients are comparably low as that in normal controls.18 In mice, it has been reported that Dnmt3a and Dnmt3b function as de novo methyltransferases during hematopoietic differentiation, and therefore have a critical role in hematopoietic stem cell self-renewal.19 In line with this notion, mutations in the aforementioned four ICF1 patients are located in the same region (patient 16: p.V726G, patient 25: p.A766P, patient 30: p.V699G and patient 45: p.R764S/p.I781KfsX23), all of which are supposed to reduce the overall stability of DNMT3B protein.20 In contrast, no decline of peripheral counts of neutrophils and thrombocytes has been observed in patients with ICF2 (two of them are >10 years old) or ICFX.
A possible T-cell defect has not been demonstrated in ICF patients as yet. However, opportunistic infections with Pneumocystis and Candida albicans occurred in several ICF patients irrespective of the group, pointing to a functional T-cell defect. In mice carrying the same missense mutations in Dnmt3b identified in ICF1 patients, massive apoptosis of T-cells was observed in the thymus. This T-cell apoptosis appears to occur a few hours after birth, as the thymocyte profiles were normal in embryonic and newborn mice. Flowcytometric analysis of CD4, CD8 and TCRβ expression revealed no developmental defect of the T-cells in the thymus of newborn mice.21 Likewise, in young ICF patients, the numbers of CD3+ T-cells, CD4+ and CD8+ T-cell subsets are normal, irrespective of the group. However, in vitro stimulation of PBMCs with PHA/IL-2 and irradiated allogeneic feeders revealed that the expansion of T-cells was reduced in all ICF1 and ICF2 cases investigated. This decreased expansion was maintained in a second round of stimulation, indicating an intrinsic T-cell defect in ICF1 and ICF2 patients, for instance a disturbed cell cycle progression. Alternatively, the reduction in T-cell numbers after stimulation could also be caused by an increased susceptibility to apoptosis of ICF1 and ICF2 T-cells, which is in line with the results obtained with splenocytes of Dnmt3b mutant mice.21
All ICF2 patients are intellectually disabled, and nearly all have a delay in development of walking and initial speech. In contrast, only half of the patients with ICF1 are intellectually disabled. Data from the Allen Brain Atlas (www.brain-map.org) shows that ZBTB24 is highly expressed in the caudate nucleus, an important part of the brain’s learning and memory system, and may explain the high incidence of intellectual disability in ICF2. In a study with transgenic mice, it has been suggested that Dnmt3b is important for the early phase of neurogenesis.22 Cerebral malformations were demonstrated in some ICF patients, belonging to both ICF1 and ICF2 patient groups.
Congenital malformations are only found in a few patients. Mouse models for ICF syndrome, either Dnmt3b knockout mice or mice carrying homozygous Dnmt3b mutations identified in ICF1 patients, demonstrated that Dnmt3b is essential for embryonic development. Dnmt3b deficiency results in embryonic lethality in mice aged E14.5–E16.5, with multiple tissue defects including ventricular septal defect.21 In agreement, congenital heart defects were also observed in three patients with DNMT3B mutations. Furthermore, two ICF2 patients carrying mutations in ZBTB24 had a cardiac defect, suggesting the effects of this gene on embryonic development.
In summary, with the identification of ZBTB24 mutations in DNMT3B mutation-negative ICF cases, it is now possible to classify ICF patients into three groups: ICF1 with mutations in DNMT3, ICF2 with mutations in ZBTB24 and ICFX patients with an unknown gene defect. In our study cohort, including two mutations not reported before, ICF1 is the most prevalent (52%), followed by ICF2 (30%). Clinically, the most striking differences are the more pronounced humoral immunodeficiency in ICF1 patients, the absence of congenital malformations in ICFX patients and the significantly higher incidence of intellectual disability in ICF2 patients. These observations on prevalence and clinical presentation may facilitate prioritization of mutation screening, and can be useful in diagnostic counseling as well. Although focus has been on the B-cell compartment, our studies indicate that the immunodeficiency in patients with ICF syndrome is not restricted to a B-cell defect, but also involves the T-cell compartment.
References
Hagleitner MM, Lankester A, Maraschio P et al: Clinical spectrum of immunodeficiency, centromeric instability and facial dysmorphism (ICF syndrome). J Med Genet 2008; 45: 93–99.
Blanco-Betancourt CE, Moncla A, Milili M et al: Defective B-cell-negative selection and terminal differentiation in the ICF syndrome. Blood 2004; 103: 2683–2690.
Pezzolo A, Prigione I, Facchetti P et al: T-cell apoptosis in ICF syndrome. J Allergy Clin Immunol 2001; 108: 310–312.
Pezzolo A, Prigione I, Chiesa S et al: A novel case of immunodeficiency, centromeric instability, and facial anomalies (the ICF syndrome): immunologic and cytogenetic studies. Haematologica 2002; 87: 329–331.
Hansen RS, Wijmenga C, Luo P et al: The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci USA 1999; 96: 1–6.
Xu GL, Bestor TH, Bourc’his D et al: Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999; 402: 1–5.
de Greef JC, Wang J, Balog J et al: Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am J Hum Genet 2011; 88: 796–804.
Chouery E, Abou-Ghoch J, Corbani S et al: A novel deletion in ZBTB24 in a Lebanese family with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Clin Genet 2012; 82: 1–5.
Jiang YL, Rigolet M, Bourc’his D et al: DNMT3B mutations and DNA methylation defect define two types of ICF syndrome. Hum Mutat 2005; 25: 56–63.
Kanariou M, Petridou E, Liatsis M et al: Age patterns of immunoglobulins G, A & M in healthy children and the influence of breast feeding and vaccination status. Pediatr Allergy Immunol 1995; 6: 24–29.
Comans-Bitter WM, de Groot R, van den Beemd R et al: Immunophenotyping of blood lymphocytes in childhood. Reference values for lymphocyte subpopulations. J Pediatr 1997; 130: 388–393.
Kloeckener-Gruissem B, Betts DR, Zankl A, Berger W, Güngör T : A new and a reclassified ICF patient without mutations in DNMT3B and its interacting proteins SUMO-1 and UBC9. Am J Med Genet 2005; 136: 31–37.
Cerbone M, Wang J, Van der Maarel SM et al: Immunodeficiency, centromeric instability, facial anomalies ICF) syndrome, due to ZBTB24 mutations, presenting with large cerebral cyst. Am J Med Genet 2012; 158A: 1–4.
van den Brand M, Flucke UE, Bult P, Weemaes CM, van Deuren M : Angiosarcoma in a patient with immunodeficiency, centromeric region instability, facial anomalies (ICF) syndrome. Am J Med Genet 2011; 155A: 622–625.
Hultén M : Selective somatic pairing and fragility at 1q12 in a boy with common variable immunodeficiency: a new syndrome. Clin Genet 1978; 14: 294–295.
Gennery AR, Slatter MA, Bredius RG et al: Hematopoietic stem cell transplantation corrects the immunologic abnormalities associated with immunodeficiency-centromeric instability-facial dysmorphism syndrome. Pediatrics 2007; 120: e1341–e1344.
Sekigawa I, Kawasaki M, Ogasawara H et al: DNA methylation: its contribution to systemic lupus erythematosus. Clin Exp Med 2006; 6: 99–106.
Zhu X, Liang J, Li F et al: Analysis of associations between the patterns of global DNA hypomethylation and expression of DNA methyltransferase in patients with systemic lupus erythematosus. Int J Dermatol 2011; 50: 697–704.
Tadokoro Y, Ema H, Okano M, Li E, Nakauchi H : De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J Exp Med 2007; 204: 715–722.
Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X : Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 2007; 449: 248–251.
Ueda Y, Okano M, Williams C et al: Roles for Dnmt3b in mammalian development: a mouse model for the ICF syndrome. Development 2006; 133: 1183–1192.
Feng J, Chang H, Li E, Fan G : Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J Neurosci Res 2005; 79: 734–746.
Tiepolo L, Maraschio P, Gimelli G et al: Multibranched chromosomes 1, 9, and 16 in a patient with combined IgA and IgE deficiency. Hum Genet 1979; 51: 1–11.
Carpenter NJ, Filipovich A, Blaese RM, Carey TL, Berkel AI : Variable immunodeficiency with abnormal condensation of the heterochromatin of chromosomes 1, 9, and 16. J Pediatr 1988; 112: 1–8.
Turleau C, Cabanis M, Girault D et al: Multibranched chromosomes in the ICF syndrome: immunodeficiency, centromeric instability, and facial anomalies. Am J Med Genet 1989; 32: 1–5.
Smeets DF, Moog U, Weemaes CMR et al: ICF syndrome: a new case and review of the literature. Hum Genet 1994; 94: 1–7.
Schuffenhauer S, Bartsch O, Stumm M et al: DNA, FISH and complementation studies in ICF syndrome: DNA hypomethylation of repetitive and single copy loci and evidence for a trans acting factor. Hum Genet 1995; 96: 562–571.
Shirohzu H, Kubota T, Kumazawa A et al: Three novel DNMT3B mutations in Japanese patients with ICF syndrome. Am J Med Genet 2002; 112: 31–37.
Björck EJ, Bui TH, Wijmenga C, Grandell U, Nordenskjöld M : Early prenatal diagnosis of the ICF syndrome. Prenat Diagn 2000; 20: 828–831.
Rigolet M, Grégoire A, Lefort G et al: Early prenatal diagnosis of ICF syndrome by mutation detection. Prenat Diagn 2007; 27: 1075–1078.
Kaya N, Al-Muhsen S, Al-Saud B et al: ICF syndrome in Saudi Arabia: immunological, cytogenetic and molecular analysis. J Clin Immunol 2011; 31: 245–252.
Brown DC, Grace E, Sumner AT, Edmunds AT, Ellis PM : ICF syndrome immunodeficiency, centromeric instability and facial anomalies): investigation of heterochromatin abnormalities and review of clinical outcome. Hum Genet 1995; 96: 1–6.
Schuetz C, Barbi G, Barth TF et al: ICF syndrome: high variability of the chromosomal phenotype and association with classical Hodgkin lymphoma. Am J Med Genet 2007; 143A: 2052–2057.
Gimelli G, Varone P, Pezzolo A, Lerone M, Pistoia V : ICF syndrome with variable expression in sibs. J Med Genet 1993; 30: 1–5.
Acknowledgements
We thank all ICF families for their participation in this study. This study was supported by the National Institute of Allergy and Infectious Diseases (NIAID: AI090135).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Weemaes, C., van Tol, M., Wang, J. et al. Heterogeneous clinical presentation in ICF syndrome: correlation with underlying gene defects. Eur J Hum Genet 21, 1219–1225 (2013). https://doi.org/10.1038/ejhg.2013.40
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.40
Keywords
This article is cited by
-
DNMT3B PWWP mutations cause hypermethylation of heterochromatin
EMBO Reports (2024)
-
Evaluation of Clinical and Immunological Alterations Associated with ICF Syndrome
Journal of Clinical Immunology (2024)
-
Enhanced CD19 activity in B cells contributes to immunodeficiency in mice deficient in the ICF syndrome gene Zbtb24
Cellular & Molecular Immunology (2023)
-
Germinal center output is sustained by HELLS-dependent DNA-methylation-maintenance in B cells
Nature Communications (2023)
-
Immunophenotype, Karyotype and Molecular Findings in a Case of ICF Syndrome
Indian Journal of Hematology and Blood Transfusion (2023)

{kind=link}
{kind=link}