
Abstract
A number of autosomal dominantly inherited disorders, such as Marfan syndrome (MFS) and Loeys–Dietz syndrome (LDS), are associated with predisposition to thoracic aortic aneurysms and dissections (TAADs). In the majority of cases, mutations in genes encoding components of the transforming growth factor-β (TGF-β) signaling pathway, such as FBN1, TGFBR1, TGFBR2 and SMAD3, underlie the disease. Recently, a familial syndromic form of TAAD with other clinical features that overlap the MFS–LDS spectrum has been described to be caused by heterozygous loss-of-function mutations in TGFB2, encoding the TGF-β2 ligand of TGF-β serine/threonine kinase receptors (TGFBRs). We analyzed the TGFB2 gene by sequencing in a cohort of 88 individuals with a Marfan-like phenotype and/or TAAD, who did not have mutations in known genes causing thoracic aortic disease. We identified the novel heterozygous c.1165dupA mutation in exon 7 of TGFB2 in three members of a family, a 51-year-old male, his brother and nephew with aortic aneurysms, cervical arterial tortuosity and/or skeletal abnormalities as well as craniofacial dysmorphisms. The 1-bp duplication causes a frameshift leading to a stable transcript with a premature stop codon after seven TGF-β2-unrelated amino acids (p.Ser389Lysfs*8). As the resulting protein is unlikely functional and by considering data from the literature, we support the notion that functional haploinsufficiency for TGF-β2 predisposes to thoracic aortic disease. Taken together, TGFB2 is a rarely mutated gene in patients with syndromic TAAD, and the clinical features of our TGFB2 mutation-positive individuals fit in the scheme of LDS, rather than MFS-related disorders.
Similar content being viewed by others


Introduction
Thoracic aortic aneurysm and dissection (TAAD) is a genetically heterogeneous disease that can occur in conjunction with several genetic syndromes, including Marfan syndrome (MFS; MIM 154700), Loeys–Dietz syndrome (LDS; MIM 609192, 608967, 610168, 613795 and 610380), vascular type of the Ehlers–Danlos syndrome (MIM 130050 and 225400) and others.1 As pathogenic mutations in various genes encoding proteins of the transforming growth factor-β (TGF-β) signaling pathway, such as FBN1, TGFBR1, TGFBR2 and SMAD3, have been identified,2, 3, 4, 5, 6, 7 dysregulated TGF-β signaling likely contributes to the pathogenesis of thoracic aortic diseases.1, 8 Indeed, several lines of evidence indicate augmented TGF-β signalling in aortic lesions in vivo.6, 9, 10 This is intriguing as many pathogenic mutations associated with TAAD have a loss-of-function effect.2, 11, 12 Recently, two studies have shown that haploinsufficiency for TGFB2 causes a familial syndrome of TAAD with additional clinical manifestations overlapping MFS and LDS.13, 14 TGFB2 encodes TGF-β2, one of three ligand isoforms that exert their cellular effects, eg, the regulation of organ-specific morphogenesis and adult tissue homeostasis by binding to heterotetrameric complexes of the TGF-β serine/threonine kinase receptors.15
Materials and methods
Patients
We obtained clinical data and blood samples from 88 individuals (both familial and sporadic cases) with a Marfan-like phenotype and/or TAAD, who were assessed by SS, MR and YvK. All individuals were negative for mutations in FBN1, TGFBR1 and TGFBR2. Sixteen patients out of these eighty-eight individuals showed clinical features overlapping with syndromic/familial forms of TAAD including vascular type of the Ehlers–Danlos syndrome (MIM 130050), aortic valve disease (MIM 109730), arterial tortuosity syndrome (MIM 208050), aneurysms-osteoarthritis syndrome (MIM 613795), familial thoracic aortic aneurysm 6 (MIM 611788) and/or familial thoracic aortic aneurysm 4 (MIM 132900). In these cases, we excluded mutations in clinically relevant, aortic disease-associated genes including COL3A1 (10 probands), ACTA2 (9 probands), SMAD3 (8 probands), SLC2A10 (3 probands), NOTCH1 (8 probands) and/or MYH11 (5 probands).
The clinical data and samples were obtained with informed consent, including consent to use the photographs in this report, under protocols approved by the Institutional Review Board.
Sequencing of TGFB2
We isolated genomic DNA from blood by standard procedures and amplified the coding region including the flanking intronic sequences of TGFB2 (exons 1–8; GenBank accession no. NM_001135599.2) from genomic DNA. Primer sequences are available on request. We directly sequenced amplicons using the ABI BigDye Terminator Sequencing Kit (Applied Biosystems, Weiterstadt, Germany) and an automated capillary sequencer (ABI 3500; Applied Biosystems, Weiterstadt, Germany). We analyzed the sequence data by the Sequence Pilot software (JSI medical systems, Kippenheim, Germany). Exon numbering of TGFB2 is according to Refseq accession number NM_001135599.2 and ensemble transcript ID ENST00000366929.
TGFB2 transcript analysis
RNA was isolated from fresh whole blood from the index patient (II-2) using the PAXgene Blood RNA System (PreAnalytics, Hombrechtikon, Switzerland) according to the manufacturer’s recommendations. First-strand cDNA was synthesized from 1 μg of RNA using the Omniscript Reverse Transcriptase Kit (Qiagen, Hilden, Germany) and gene-specific oligonucleotides. Of a total of 20 μl of volume from the reaction, 1 μl was used as template to amplify transcripts by using primer pairs spanning different exon–exon junctions in nested PCR reactions. Amplicons were directly sequenced or cloned into pCR2.1 TOPO TA Cloning Vector (Invitrogen, Darmstadt, Germany). For semi-quantitative analysis, 34 Escherichia coli clones were subjected to colony PCR, and PCR products from individual clones were sequenced.
Database
Mutation data (TGFB2 c.1165dupA) were submitted to Leiden Open Variation Database (LOVD) for TGFB2 at http://www.LOVD.nl/TGFB2.
Results
Mutation analysis of TGFB2
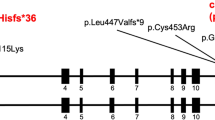
We sequenced the TGFB2 gene in a cohort of 88 individuals with a Marfan-like phenotype and/or TAAD and detected a single sequence alteration: the heterozygous mutation c.1165dupA in the penultimate codon of exon 7 (Figure 1a) in a 51-year-old male with TAAD and a phenotype resembling MFS (Figure 2a). The 1-bp duplication likely leads to a frameshift and introduction of a premature stop codon after seven TGF-β2-unrelated amino acids (p.Ser389Lysfs*8; Figure 1b). By RNA analysis, we demonstrated expression of both the wild-type and the mutated TGFB2 allele in blood cells of the index patient (Figure 1c); semi-quantitative evaluation suggested 59% and 41% of wild-type and mutant TGFB2 transcripts, respectively (data not shown). We sequenced exon 7 of TGFB2 in four additional family members of the index patient and identified the c.1165dupA mutation in one of his two affected brothers (II-4 in Figure 2b; II-6 was not available for testing) and his nephew (Figure 2b, III-2), who showed aortic aneurysm and/or a Marfan-like phenotype. We could exclude the mutation in the healthy daughter (III-1 in Figure 2b) and the healthy brother (II-5 in Figure 2b) of the index patient. In summary, the TGFB2 mutation co-segregates with the disease phenotype in the family.

Molecular aspects and consequences of the TGFB2 c.1165dupA mutation. (a) Part of the sequence electropherogram of TGFB2 exon 7–intron 7 (gDNA, genomic DNA). The index patient is heterozygous for the 1-bp duplication c.1165dupA in TGFB2 (indicated by an arrow, ↓). The sequence of the wild-type and mutated allele is given above the electropherogram; the exon–intron boundary and encoded amino acids (one-letter-code) are indicated. (b) Schematic presentation of TGFB2 messenger RNA aligned with protein domains of the TGF-β2 proprotein. The c.1165dupA mutation is predicted to cause a frameshift leading to a premature stop codon (PTC) in TGFB2 exon 8 and a hypothetically COOH-terminal truncated protein (p.Ser389Lysfs*8). Protein domains are indicated according to Simple Modular Architecture Research Tool (http://smart.embl-heidelberg.de/). ATG, start codon; TAA, stop codon; UTR, untranslated region; aa, amino acid. (c) Transcript analysis: partial sequence electropherogram of TGFB2 exon 7–exon 8 junction obtained from cDNA (complementary DNA). The index patient is heterozygous for the TGFB2 c.1165dupA mutation (indicated by an arrow, ↓). The sequence of the wild-type (WT) and mutated (MUT) allele is given under the electropherogram; the exon 7–exon 8 junction and the PTC (STOP) is shown.

Photographs and family pedigree. (a) Photographs of the index patient. Note the cerebral arterial tortuosity, the skeletal abnormalities including pes planus and retrognathia, but negative wrist and thumb signs and no restriction of elbow extension. The patient shows postoperative scars derived from aortic aneurysm surgery and former surgical operations. (b) The three-generation pedigree shows individuals with clinical manifestations suggestive of a MFS-LDS spectrum disorder (▪). The index patient (II-2) is indicated by an arrow. Three affected individuals (II-2, II-4 and III-2) share the mutated TGFB2 allele. Material from the deceased father (I-1) of the brothers for TGFB2 sequencing was not available; one affected brother (II-6) denied genetic testing. Plus sign (+) in symbols, mutation present; minus sign (−) in symbols, mutation absent; gray bars above symbols, DNA available.
Clinical data of family members with the TGFB2 c.1165dupA mutation
The index patient II-2 (Figure 2b) was clinically evaluated at age 51 years. His height was 191 cm and weight 85 kg. He had mild scoliosis, pes planus and a positive ratio of arm span and body height. Craniofacial dysmorphism included malar hypoplasia, prominent uvula, elongated face and retrognathia (Figure 2a). He already had undergone several surgical operations including testicular torsion repair, inguinal hernia surgery, appendectomy and sigma resection after acute diverticulitis. Magnetic resonance angiography revealed profound cerebral and cervical arterial tortuosity (Figure 2a). After we diagnosed aortic insufficiency due to an aneurysm with a diameter of 51 mm, patient II-2 underwent aortic aneurysm surgery. Based on these clinical manifestations, we made the diagnosis of a syndromic TAAD disorder. The brother of patient II-2, II-4 in Figure 2b, whose height was 186 cm and weight 104 kg, presented with arterial tortuosity, mild scoliosis, joint hyperflexibility, retrognathia and a high arched palate at age 47 years. He had undergone aortic aneurysm surgery because of an aneurysm with a diameter of 58 mm and inguinal hernia surgery. Another brother of the index patient, II-6 in Figure 2b, also had undergone aortic aneurysm surgery, and he showed clinical features strongly suggestive of a MFS-related disorder. He denied genetic testing. The index patient’s nephew, III-2 in Figure 2b, was clinically evaluated at the age of 13 years. He had scoliosis, pectus deformity, joint hyperflexibility and club foot, which were indicative for a syndromic TAAD disorder. The father I-1 (Figure 2b) of the index patient and his brothers died at the age of 52 years after acute aortic dissection.
Discussion
We report a familial syndromic form of thoracic aortic aneurysm because of a heterozygous 1-bp duplication (c.1165dupA) in proximity of the last exon–intron boundary in TGFB2 leading to a frameshift and a PTC (p.Ser389Lysfs*8). Because the PTC is located in the last TGFB2 exon (Figure 1b), the mutant transcript is not expected to be degraded by nonsense-mediated mRNA decay.16 Indeed, we detected expression of both wild-type and mutant TGFB2 transcripts. However, we propose that the mutated TGF-β2 p.Ser389Lysfs*8 protein is not functional, either because of a severely compromised reactivity of the growth-factor domain or because of misfolding and degradation before secretion. In line with this, normal levels of mutant TGFB2 transcripts but reduced proprotein levels have been detected in smooth muscle cells and fibroblasts from patients with the frameshift mutation c.1106_1110delACAAT (p.Tyr369Cysfs*26) in TGFB2 (RefSeq NM_001135599.2, NP_001129071.1); this alteration was originally described as c.1021_1025delTACAA (p.Tyr341Cysfs*25) (TGFB2 RefSeq NM_003238.3, NP_003229.1).13 Considering these consequences, our finding of the c.1165dupA mutation in a three-generation family supports the idea that functional haploinsufficiency of TGF-β2 causes a cellular compensatory overshoot leading to augmented TGF-β signalling in MFS-LDS spectrum disorders.17 On the other hand, we cannot exclude that, due to structural changes, the p.Tyr341Cysfs*25 mutation enhances TGF-β receptor affinity resulting in increased TGF-β signaling.
Auxiliary receptor TGFBR3/betaglycan is required for cellular responsiveness to TGF-β2.18, 19 Non-synonymous variants [c.334T>A (p.(W112R)), c.2368A>T (p.(I790F))] have been identified in TGFBR3 in patients with familial intracranial aneurysms,20 which strongly suggests a role of TGF-β2-induced signaling in the molecular pathogenesis of aneurysms.
Interestingly, a mutation in TGFB3 encoding the TGF-β3 ligand is associated with a syndrome with few features of MFS and LDS, but no vascular findings suggesting a pathophysiology distinct from that caused by excess TGF-β signaling.21
Up to date, 12 independent TGFB2 mutations in 34 individuals have been reported (Table 1).13, 14 In our study, the three subjects with the c.1165dupA mutation in TGFB2 share clinical features with other autosomal dominant aortic aneurysm syndromes. Among these features, aortic aneurysm, scoliosis, pectus deformity, joint hyperflexibility, pes planus, retrognathia, high arched palate and hernia occur both in MFS and LDS.22 Conversely, arterial tortuosity and club feet are manifestations, which are only seen in LDS. Moreover, the index patient did not present with reduced elbow extension or positive wrist/thumb signs, which are common manifestations of MFS. Most notably, the patients in this report did not exhibit ectopia lentis, which occurs in MFS but not in LDS. Similarly, Lindsay et al14 did not observe ectopia lentis in any of the 15 individuals with TGFB2 mutations (Table 1),14 and Boileau et al13 described only a single patient among 13 individuals with TGFB2 mutations who exhibited some sort of ‘minor ectopia lentis’, which they stated was ‘not recognized as lens dislocation in the USA.’13 Thus, the clinical presentation of the three affected male relatives described here strengthens the notion that mutations in TGFB2 are associated with an LDS rather than a MFS phenotype.14
We identified only one mutation carrier in a cohort of 88 individuals (1.1%) with a phenotype within the MFS-LDS spectrum. Similarly, TGFB2 mutations were identified in 4 out of 409 probands (1%) including both familial and sporadic cases with thoracic aortic disease.13 In contrast, Lindsay et al14 found a relative high frequency of TGFB2 mutations (in 6 out of 86 individuals, 7.0%), which may be the result of testing a cohort of probands with phenotypic features within the LDS spectrum.14 Notably, only two patients with MFS/LDS-related disorders in combination with developmental delay and heterozygous de novo microdeletions (6.5 Mb and 3.5 Mb) including the TGFB2 gene have been described;14 however, more detailed copy number analysis may uncover further TGFB2 mutations in patients with thoracic aortic disease. Taken together, sequence analysis identified pathogenic TGFB2 lesions in about 2% of patients with thoracic aortic disease, which is a rather low frequency compared with the three major TGF-β vasculopathy-associated genes FBN1, TGFBR1 and TGFBR2. However, in view of the high risk for aortic aneurysm and dissection associated with TGFB2-mutations, molecular testing of this gene is crucial. Therefore, we suggest that a clinical presentation including thoracic aortic disease, arterial tortuosity, skeletal abnormalities and hernia but not ectopia lentis is indicative for mutation testing of TGFB2. Nevertheless, there are still many mutation-negative individuals with thoracic aortic disease indicating further genetic heterogeneity and additional genetic studies are necessary to recognize all details of the pathogenesis underlying this disorder.
Accession codes
References
Milewicz DM, Guo DC, Tran-Fadulu V et al: Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet 2008; 9: 283–302.
Loeys BL, Schwarze U, Holm T et al: Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 2006; 355: 788–798.
van de Laar IM, Oldenburg RA, Pals G et al: Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet 2011; 43: 121–126.
Regalado ES, Guo DC, Villamizar C et al: Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res 2011; 109: 680–686.
Dietz HC, Cutting GR, Pyeritz RE et al: Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991; 352: 337–339.
Loeys BL, Chen J, Neptune ER et al: A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 2005; 37: 275–281.
Mizuguchi T, Collod-Beroud G, Akiyama T et al: Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet 2004; 36: 855–860.
Pardali E, Goumans MJ, ten Dijke P : Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol 2010; 20: 556–567.
Inamoto S, Kwartler CS, Lafont AL et al: TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc Res 2010; 88: 520–529.
Gomez D, Al Haj Zen A, Borges LF et al: Syndromic and non-syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J Pathol 2009; 218: 131–142.
Akhurst RJ : The paradoxical TGF-beta vasculopathies. Nat Genet 2012; 44: 838–839.
Langlois D, Hneino M, Bouazza L et al: Conditional inactivation of TGF-beta type II receptor in smooth muscle cells and epicardium causes lethal aortic and cardiac defects. Transgenic Res 2010; 19: 1069–1082.
Boileau C, Guo DC, Hanna N et al: TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet 2012; 44: 916–921.
Lindsay ME, Schepers D, Bolar NA et al: Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet 2012; 44: 922–927.
Moustakas A, Heldin CH : The regulation of TGFbeta signal transduction. Development 2009; 136: 3699–3714.
Silva AL, Romao L : The mammalian nonsense-mediated mRNA decay pathway: to decay or not to decay! Which players make the decision? FEBS Lett 2009; 583: 499–505.
Lindsay ME, Dietz HC : Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 2011; 473: 308–316.
Lopez-Casillas F, Wrana JL, Massague J : Betaglycan presents ligand to the TGF beta signaling receptor. Cell 1993; 73: 1435–1444.
Rodriguez C, Chen F, Weinberg RA, Lodish HF : Cooperative binding of transforming growth factor (TGF)-beta 2 to the types I and II TGF-beta receptors. J Biol Chem 1995; 270: 15919–15922.
Santiago-Sim T, Mathew-Joseph S, Pannu H et al: Sequencing of TGF-beta pathway genes in familial cases of intracranial aneurysm. Stroke 2009; 40: 1604–1611.
Rienhoff HY Jr, Yeo CY, Morissette R et al: A mutation in TGFB3 associated with a syndrome of low muscle mass, growth retardation, distal arthrogryposis and clinical features overlapping with marfan and Loeys-Dietz syndrome. Am J Med Genet A 2013; 161: 2040–2046.
von Kodolitsch Y, Robinson PN : Marfan syndrome: an update of genetics, medical and surgical management. Heart 2007; 93: 755–760.
Acknowledgements
We are grateful to the family members for their interest and participation in this study. The results summarized here form part of the MD thesis of Ruth Leutermann at the University of Hamburg. We thank Inka Jantke for excellent technical assistance. This work was supported by grants of the Deutsche Forschungsgemeinschaft (KU 1240/6–1 to KK and RO 3660/1–1 to GR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Leutermann, R., Sheikhzadeh, S., Brockstädt, L. et al. A 1-bp duplication in TGFB2 in three family members with a syndromic form of thoracic aortic aneurysm. Eur J Hum Genet 22, 944–948 (2014). https://doi.org/10.1038/ejhg.2013.252
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.252
Keywords
This article is cited by
-
Association of MLL3 and TGF-β signaling gene polymorphisms with the susceptibility and prognostic outcomes of Stanford type B aortic dissection
BMC Cardiovascular Disorders (2023)
-
A missense TGFB2 variant p.(Arg320Cys) causes a paradoxical and striking increase in aortic TGFB1/2 expression
European Journal of Human Genetics (2017)
-
Further delineation of Loeys-Dietz syndrome type 4 in a family with mild vascular involvement and a TGFB2 splicing mutation
BMC Medical Genetics (2014)
