
Abstract
Mucolipidosis (ML) II and ML IIIα/β are allelic autosomal recessive metabolic disorders due to mutations in GNPTAB. The gene encodes the enzyme UDP-GlcNAc-1-phosphotransferase (GNPT), which is critical to proper trafficking of lysosomal acid hydrolases. The ML phenotypic spectrum is dichotomous. Criteria set for defining ML II and ML IIIα/β are inclusive for all but the few patients with phenotypes that span the archetypes. Clinical and biochemical findings of the ‘intermediate’ ML in eight patients with the c.10A>C missense mutation in GNPTAB are presented to define this intermediate ML and provide a broader insight into ML pathogenesis. Extensive clinical information, including radiographic examinations at various ages, was obtained from a detailed study of all patients. GNPTAB was sequenced in probands and parents. GNPT activity was measured and cathepsin D sorting assays were performed in fibroblasts. Intermediate ML patients who share the c.10A>C/p.K4Q mutation in GNPTAB demonstrate a distinct, consistent phenotype similar to ML II in physical and radiographic features and to ML IIIα/β in psychomotor development and life expectancy. GNPT activity is reduced to 7–12% but the majority of newly synthesized cathepsin D remains intracellular. The GNPTAB c.10A>C/p.K4Q missense allele results in an intermediate ML II/III with distinct clinical and biochemical characteristics. This delineation strengthens the utility of the discontinuous genotype–phenotype correlation in ML II and ML IIIα/β and prompts additional studies on the tissue-specific pathogenesis in GNPT-deficient ML.
Similar content being viewed by others

Introduction
Mucolipidosis II (ML II; I–cell disease: MIM 252500) and mucolipidosis IIIα/β (ML III; pseudo-Hurler-polydystrophy; MIM 252600) are allelic autosomal recessive metabolic disorders directly correlated with homozygous or compound heterozygous mutant GNPTAB genotypes. The GNPTAB gene encodes the α and β polypeptides in the hexameric enzyme UDP-GlcNAc-1-phosphotransferase (GNPT: IUBMB no. 2.7.8.17). The γ polypeptide is encoded by the GNPTG gene. The enzyme complex catalyzes the first step in the biosynthesis of the mannose-6-phosphate (M6P) recognition marker in the oligomannosyl-type N-linked glycans in lysosomal acid hydrolases. Without the M6P marker most soluble hydrolases are not targeted to the lysosomal compartment in connective tissues, resulting in impaired lysosomal degradation.
GNPTAB genotypes, either homozygous or compound heterozygous for mutant alleles with ‘amorph’ or ‘null’ effect (frameshift and nonsense mutations), result in the early-onset, severe ML II. The later-onset ML IIIα/β with prolonged course is the morbid result in patients with at least one ‘hypomorph effect’ allele (missense and most splice mutations) in the GNPTAB mutant genotype. The phenotypic spectrum of patients with GNPT-deficiency disorders is dichotomous rather than continuously variable. ML II and ML IIIα/β are phenotypically distinguishable. Within the largest published cohort of ML patients, the phenotype of only a few subjects straddled the criteria set for defining ML II and ML IIIα/β.1
The purpose of this report is to present and discuss the clinical and biochemical findings of the ‘intermediate’ ML in eight patients with the c.10A>C missense mutation in exon 1 in their compound heterozygous GNPTAB genotype. The amino acid lysine is replaced with glutamine, p.K4Q (designated K4Q hereafter). The name ‘Webb type ML’ is proposed following the explicit permission for use of the surname of one of the families and honoring all participating families and patients in this study. The findings corroborate the overall genotype–phenotype correlation, underscore the predictive power of mutation screening and illustrate the potential for generating heuristic hypotheses on the pathogenesis of ML.
Subjects and methods
Probands 1, 2 and 3 are isolated patients in three unrelated families. Only patient 3, who died at the age of 23 6/12 years, was never examined by at least one of the authors. However, the wealth of clinical data made available by the mother prompted inclusion in the core group of subjects reported earlier and in this study.1 The postmortem study of patient 3 was recently published as the report of autopsy findings in a patient with ML IIIα/β.2 Patients 4–8 are siblings. The earlier part of the disorder’s course in the older siblings was reported by Kozlowski et al.3 All sibs were included in the recent report by David-Vizcarra et al,4 who recognized the affected as examples of ML intermediate between ML II and ML IIIαβ. The parents in each of the four families were non-consanguineous.
In addition to plasma samples and fibroblast cell lines representing the eight subjects, cultured fibroblasts from two additional patients, one homozygous and one compound heterozygous for the K4Q missense mutation, were obtained from NIGMS Human Genetic Cell Repository at Coriell Institute (Camden, NJ, USA) and included in the biochemical studies. No clinical information except the diagnosis of ML III was available. The mutant genotypes in patients and cell lines are presented in Table 1.
The activity of selected lysosomal acid hydrolases was assayed in plasma and cultured fibroblasts according to the methods described by Thomas et al.5 Metabolic labeling and sorting of the lysosomal hydrolase cathepsin D in control and patient fibroblasts were performed as described previously.6, 7 Briefly, cell monolayers at about 60–80% confluency were washed twice with phosphate-buffered saline and methionine starved using cysteine/methionine-free Dulbecco’s modified Eagle’s medium for 30 min, followed by a pulse label for 1 h with 1 ml of 1 mCi/ml Tran35S-label (MP Biomedicals, Solon, OH, USA) in the same media. Excess unlabeled methionine (10 mM final concentration) was added to initiate a 4-h chase. The amount of cathepsin D sorted was determined by overnight immunoprecipitation of equivalent aliquots of the lysed cell and media high-speed supernatants at 4 °C with rabbit anti-human cathepsin D antiserum and protein A-Sepharose beads. After five washes with buffer containing 1 M KCl and 1% Triton X-100, immunoprecipitates were subjected to 10% SDS-PAGE under nonreducing conditions. Gels were dried and exposed on an autoradiography film. Quantification of the various bands was performed using Image J software. The percentage of cathepsin D that remains intracellular was calculated as the densitometric ratio of the intracellular forms (both intermediate and mature) to the sum of the intracellular and secreted (precursor) forms.
GlcNAc-1-phosphotransferase activity was measured in microsomal preparations from fibroblasts as described earlier.8, 9 Briefly, cells were lysed by sonication in Tris buffer pH 7.5 containing 0.25 M sucrose and microsomes prepared by ultracentrifugation. Reactions using 10 mM α-methylmannoside as an acceptor and 3H-UDP-GlcNAc (along with 200 μ M ‘cold’ UDP-GlcNAc) as substrate were conducted in the following reaction buffer: 50 mM Tris pH 7.5, 0.3% Triton X-100, 50 mM N-acetylglucosamine, 1 mM DTT, 20 mg/ml bovine serum albumin, 10 mM sodium molybdate, 1 mM MgCl2, and 1 mM MnCl2. Activity units are calculated as pmoles of 3H GlcNAc transferred per hour. Protein concentration was measured using the micro BCA protein assay kit (Pierce, Rockford, IL, USA).
Screening for the GNPTAB mutations by PCR and sequencing protocols was performed according to the methods reported earlier.1
Results
Clinical phenotype
Patient 1
Patient 1 was delivered at term after an uncomplicated pregnancy (panels A1 and A2 in Figure 1). Size was appropriate for gestational age (Table 2). Early features included plagiocephaly treated with helmeting, kyphosis noted at approximately 9 months and regurgitant cardiac valves confirmed by echocardiogram at 12 months. Skeletal survey was performed at 20 months when a decreased range of motion in the shoulders prohibited lifting the arms above the head. The radiographs showed features of dysostosis multiplex (DM) that included 33° kyphosis, varus angulation of the proximal humeri, bowed, dysplastic radii and ulnae, proximal and distal tapering of the widened metacarpals, and marked coxa valga and subluxation of the hips. The diagnosis of ML II was assigned at 22 months. While anthropometric measurements were normal in the first year of life, growth failure was apparent from his second year. At 1 year, length was 76 cm (50th percentile). The maximal height of 80.5 cm (mean for a 16-month-old male) was recorded at 7.5 years. Speech and expressive language development were normal but unaided walking was not achieved until 20 months. Cognitive development was delayed. Formal assessment at 12 11/12 years showed a global intellectual ability score of 39, comparable to an age equivalent of 5 5/12 years. Obstructive sleep apnea was treated with continuous positive airway pressure at night, beginning before 5 years. Recurrent infections led to one hospitalization for pneumonia and three sets of myringotomy tubes by 8 years. Several surgical attempts were made to stabilize his progressive thoracolumbar kyphosis, with operations at 3, 6, and 10 years. Surgery for relief of bilateral carpal tunnel syndrome was performed at 7 years. Postoperative extubation was consistently difficult. Prolonged ventilator support with slow weaning was required. When examined at 12 7/12 years progressive joint contractures had decreased measured height to 78.7 cm (mean for a 15-month-old male), weight was 15 kg (mean for a 3-year-old male), and head circumference 51.5 cm (mean for a 6-year-old male). He wore glasses for nearsightedness. Dysmorphic features included flat and coarse facies, ridged palate, thickened gums, broad and short hands held in ulnar deviation, broad and camptomelic fingers. There was a small umbilical hernia and diastasis of the straight abdominal muscles without organomegaly. Despite joint restriction, the boy was mobile, interactive and actively playful. He conversed appropriately with hoarse voice.

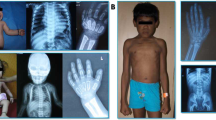
GNPTAB c.10A>C/p.K4Q ML clinical features in three unrelated patients: short stature, normocephaly, coarse facies, limited mobility in all joints, short and broad hands with fingers postured in ulnar deviation. A1 and A2: Patient 1 at 8 and 12 years; B: Patient 2 at 6 years; C1 and C2: Patient 3 at 8 and 20 years.
Patient 2
Patient 2 was delivered by cesarean section for transverse lie following a pregnancy complicated by hyperemesis in the first trimester (panel B in Figure 1). Ultrasound examination at 18 weeks gestation was normal. Neonatal anthropometrics were normal (Table 2). Features noticed on the first day of life included unusual facies with micrognathia, contractures at the knees and hips but hypermobile wrists, slender fingers and narrow feet. Slow weight gain in infancy was improved temporarily when breastfeeding was supplemented with bottle-feeding. Impaired growth was apparent by 22 months. Speech developed normally. He had significant gross motor delay, persistent truncal hypotonia and mild thoracolumbar kyphoscoliosis noticed at 12 months. He did not sit unassisted until 18 months and walked at 30 months. At 33 months, he did not have organomegaly, corneal clouding or gingival swelling, but lysosomal storage disease was suspected based on his coarse facies, short stature, and skeletal changes. Testing revealed increased activity of multiple lysosomal enzymes in plasma. The initial diagnosis of ML II was assigned, but by age 40 months the overall clinical phenotype was more in support of intermediate ML and called ‘type II/III’. Examination at 10 2/12 years showed an alert and conversant, ambulatory boy with stature 86.5 cm (mean for a 23-month-old male), weight 16.4 kg (mean for a 12-month-old male), and head circumference 51.5 cm (mean for a 6-year-old male). Relevant findings included normocephaly, coarse facies, gingival hypertrophy, narrow thorax, soft ejection murmur, mildly prominent abdomen without organomegaly, umbilical hernia, thoracolumbar kyphoscoliosis, severely limited range of motion in all joints, short claw-like hands in ulnar deviation, and broad camptomelic fingers.
Patient 3
Patient 3 was the third of three sons born to a Japanese father and Caucasian mother (panels C1 and C2 in Figure 1). Pregnancy was uncomplicated until 4 weeks prior to the expected delivery date when rupture of membranes and labor led to vacuum-assisted vaginal delivery. Size was appropriate for gestational age (Table 2). By 9 months, parents regarded his development to be slower than that in his healthy siblings and noted differing physical features, including proptotic eyes, short neck, relatively large head, narrow shoulders, and protruding abdomen. Speech was late (first words at 15 months) and difficult to understand. He achieved unaided walking at 18 months despite stiff hips and knees. By 23 months, he had limited range of motion at the shoulders, wrists and fingers, and thoracolumbar kyphoscoliosis. ML II was first suspected at 26 months; ML III was his diagnosis at 29 months. Minimal statural growth occurred after age 3 years. He had speech therapy and was in special education classes throughout the years of schooling. Testing of academic achievement at 14 years revealed a school performance at the third to fourth grade level. At 11 years, he had mild thickening of aortic and mitral valves, mitral regurgitation and left ventricular hypertrophy. Cardiac and pulmonary function worsened in the second decade. At 21 years, the left ventricular ejection fraction was below 60% and associated with mild to moderate regurgitation at all cardiac valves. He had severe sleep apnea by mid-teens and required continuous supplemental oxygen by 22 years. He died at 23 years. Autopsy confirmed severe dilated hypertrophic cardiomyopathy.2
Patients 4–8: common features in sibling patients
Five of seven children born to healthy parents were affected (Figure 2; Table 2). The two surviving affected siblings (patients 5 and 8) are ages 28 and 15 years. Patients 4, 6, and 7 died at 21, 24, and 14 years, respectively. Placental abruption at term complicated one pregnancy. The third affected child, a male, was diagnosed prenatally and had an unaffected female twin. No ultrasound abnormalities had been detected in either fetus. Kyphosis and dislocated hips were diagnosed in the firstborn child (patient 4) when she was 9 months old. Dysmorphic facial features and joint contractures were subsequently recognized in infancy in each of the affected siblings. The diagnosis of ML II was made simultaneously in the two oldest children at 26 months and 7 months. All of the children were of appropriate size for gestational age, but statural growth slowed from infancy and ceased in early childhood. Receptive and expressive language development occurred at a near normal pace but neuromotor development was delayed. Ambulation was not achieved until age 7 years in patient 4, who had severe congenital flexion contractures at the knees. Patient 7 maintained independent ambulation only briefly. Cognitive development was impaired, and all participated in special education schooling. None truly mastered reading but could copy letters and words. Recurrent bouts of otitis contributed to conductive hearing loss and led to repeated sets of myringotomy tubes in all. All patients had surgery for carpal tunnel syndrome. Systolic heart murmurs were present from early childhood. Echocardiography revealed slow thickening and gradually worsening incompetence of the mitral and aortic valves. Later in the clinical course, enlargement of the left ventricular wall and dilatation of the left atrium and ventricle were noticed. From infancy, breathing was louder, more superficial and faster than normal. Adenoidectomy was performed in some. Sleep apnea, obstructive and central, was treated with continuous positive airway pressure. There was limited development of secondary sexual characteristics; only one of the females ever had menses. Disease progression was characterized by severe bone and joint pain even when at rest. Periodic intravenous infusion of biphosphonate provided transient pain relief. All patients developed radiographic and neurologic signs of narrowing of the spinal canal. Although disease progression was apparent in every system, the morbid consequences of cord compression dominated end-of-life issues.

GNPTAB c.10A>C/p.K4Q ML clinical features in five siblings: Similar findings as in Figure 1 are shown early and late in disorder’s natural course. D: Patient 4 at 21.5 years; E: Patient 5 at 20 years; F: Patient 6 at 17.5 years; G: Patient 7 at 7.5 years; H: Patient 8 at 6 years.
The clinical diagnosis of intermediate mucolipidosis (ML II/III) was reached in the two prior reports that included clinical and radiological data from affected members of this family.3, 4
The radiographic phenotype
The osteochondrodysplasia (OCD) documented radiographically in this group of ML patients fits within the concept of DM defined and illustrated by Spranger et al.10 The skeletal radiographs in this group of patients at various ages show the remarkably consistent DM that is congruent with ML II and significantly different from the ML IIIα/β radiologic phenotype (Table 3; Figures 3 and 4).1, 3, 4, 10, 11

Lateral radiographs of spinal columns in eight patients with GNPTAB c.10A>C/p.K4Q ML compared with one ML II and two ML III α/β patients. Thoracolumbar vertebral bodies remain significantly foreshortened with concave frontal and convex upper/lower margins; delayed and deficient ossification. Similar findings are seen in ML II. In ML IIIα/β vertebral bodies are oblong, and show mild platyspondyly and irregular margins higher anteriorly than posteriorly. In all types of ML ossification is most deficient in either T12 or L1 or in both, often at the top of thoracolumbar kyphoscoliosis. A. Patient 1 at 8 years; B: Patient 2 at 6 years; C: Patient 3 at 9 years; D: Patient 4 at 19 years; E: Patient 5 at 19 years; F: Patient 6 at 18 years; G: Patient 7 at 7 years; H: Patient 8 at 4 years; ML II: female ML II patient at 4 years; ML III-1: male with ML III α/β at 18 years; ML-III-2: female with ML III α/β at 13 years.

Hand radiographs in GNPTAB c.10A>C/p.K4Q ML patients: comparison with ML II and ML IIIα/β. DM in the hands is similar to that in ML II and distinctly more severe than in ML IIIα/β. Figures show shortening and diaphyseal widening in all tubular hand bones; severely delayed and deficient ossification of epiphyses, carpal bones and the distal ulnar and radial epiphyses; and apparent regression of mid-diaphyseal widening in older patients concomitant with progressive generalized osteopenia. A: Patient 1 at 8 years; B: Patient 2 at 4 years; C: Patient 3 at 9 years; D: Patient 4 at 9 years; E: Patient 5 at 19 years; H: Patient 8 at 6 years; ML II: female with ML II at 4 years; ML III-2: female with ML III α/β at 13 years.
In the lateral skull films available from late childhood only the sella turcica has an elongated shape, whereas in age-matched and older ML IIIα/β patients it maintains the normal configuration. The thoracolumbar vertebrae demonstrate a much foreshortened and near ovoid shape with convex upper and lower borders, and concave anterior and posterior irregular rims. At least one vertebra, either T12 and/or L1 at the top of the kyphotic spine deformation appears more hypoplastic, insufficiently ossified and often triangular (Figure 3). The latter feature is consistently present in ML II, but absent in some ML IIIα/β patients. In the anteroposterior thoracic films, the ribs are considerably widened in the lateral and anterior segments, but abnormally narrow near the vertebral insertion (not shown). The pelvis is most obviously osteopenic with severely slanted acetabular roofs, narrow basilar portions of the ilia and relatively large and elongated ischial and pubic bones, features consistently present in ML II and also ML IIIα/β. All long bones are short and poorly tubulated with thick cortices and small, coarsely trabeculated epiphyses. The knee epiphyses are small and almost rectangular. Ossification of the lateral portion of the tibial submetaphyseal zone is overmodeled. Changes in the hands and wrists (Figure 4) fit into the severe side of the DM spectrum. All epiphyses in the tubular hand bones and the carpal bones remain small and irregular. The metacarpal diaphyses are excessively widened and ends are constricted. While nearly congruent with the changes in ML II, the hand skeleton in these intermediate ML patients differs significantly from the much milder OCD in ML IIIα/β hand bones (Figure 4). In the longest surviving patients, the radiographic findings are altered by the worsening osteopenia.
Biochemical and molecular results
Acid glycosidase levels in serum and cells
The activity of total β-D-hexosaminidase, α-L-fucosidase and β-glucuronidase was significantly increased in plasma and decreased in cultured fibroblasts derived from patients. These results overlap with the ranges of hydrolase activity observed in ML II and ML IIIα/β. These findings do not contribute to distinguishing the GNPTAB c.10A>C/p.K4Q ML patients from either of the archetype GNPT-deficient disorders.1, 12, 13
Assay of UDP-GlcNAc 1-phosphotransferase (GNPT)
The results of the GNPT assay are presented in Table 4. The activity of the phosphotransferase towards the substrate α-methylmannoside is reduced to a range of 7–12% in the GNPTAB c.10A>C/p.K4Q ML patients, about half of that in the K4Q homozygous patient fibroblasts (GM00113), which is to be expected.14 The significantly higher GNPT activity in the K4Q samples compared with classic ML II supports the distinct nature of the GNPTAB c.10A>C/p.K4Q ML.
Sorting efficiency of cathepsin D
The phenomenon of missorting of immature lysosomal enzymes and their appearance outside the cell was studied in nascent cathepsin D by pulse-chase immunoprecipitation experiments. Cathepsin D is a highly mannose phosphorylated lysosomal protease that is hypersecreted from the cell when mannose phosphorylation is impaired. The pulse/chase assays thus provide an opportunity to gauge the amount of newly synthesized cathepsin D that remains intracellular versus that secreted into the media. Whereas the level of intracellular cathepsin D in fibroblasts from either controls or obligate heterozygous parents exceeds the normal 90% (Figure 5), ML II-type ‘I-cells’ (GM01586) retain only about 20% of newly synthesized cathepsin D. The fibroblasts from our patient 2 and the homozygous K4Q cells (GM0113) exhibit surprising levels of intracellular cathepsin D (81% and 65%, respectively). The comparatively reduced percentage in the GM00113 cells is attributed to the late passage number and consequent slow-growing nature of this culture. Greater heterogeneity in all forms of cathepsin D was detected in patient 2 and GM00113, which may reflect altered trafficking and/or processing of this hydrolase within the secretory pathway.

Sorting of newly synthesized cathepsin D in control and ML fibroblasts GM01586 (ML II), patient 2 (K4Q compound heterozygote), and GM00113 (K4Q homozygote). Autoradiographs of 35S-labeled immunoprecipitates of cell and secreted cathepsin D are shown. The percentage of intracellular enzyme is determined by dividing the amount of cell-associated cathepsin D by the total cathepsin D (cell-associated and secreted).
GNPTAB mutation screening
The GNPTAB mutation analysis data in patients 1–3 and 8 were previously reported.1 Table 1 also lists the mutant GNPTAB genotypes for the two fibroblast lines obtained from the Coriell Institute.
Discussion
Of the 61 probands included in the report by Cathey et al,1 the clinical features in only a few patients with GNPT-deficient ML straddled the boundaries of the variability spectra set for delineation of ML II and of ML IIIα/β.1 Upon referral for GNPTAB mutation screening, all had been diagnosed as ‘intermediate ML’ or ‘ML II/III’. The majority of these ‘intermediate’ patients was found to have a compound heterozygous mutant genotype that consistently included c.10A>C/p.K4Q missense mutation in exon 1 and a frameshift mutation in a downstream exon (Table 1). This group of ML patients has a distinct and remarkably consistent phenotype that shares physical (Figures 1 and 2) and radiographic (Figures 3 and 4) features with ML II, but more closely resembles the ML IIIα/β phenotype regarding life expectancy, psychomotor development and degree of intellectual disability.
The GNPTAB missense (MS) mutation c.10A>C/p.K4Q causes a unique intermediate type ML. The two cytosolic domains of GNPTAB at the N- and C-termini contain motifs that mediate the export or trafficking of the enzyme through the endoplasmic reticulum and Golgi. The importance of the N-terminal domain has been highlighted by recent work from Franke et al15 showing that the lysine residue at position 4—the same residue mutated in the patients described in this report—affects a neighboring dileucine sequence responsible for the export of the GlcNAc-1-phosphotransferase αβ precursor from the endoplasmic reticulum.15 The possibility that other mutations may be found to result in an identical phenotype cannot be excluded. It remains to be seen whether other mutations within the N-terminal domain, or perhaps the C-terminal cytosolic domain, are associated with the unique clinical and biochemical features described in the patients reported here. This distinct ML subtype may be associated with other mutations that directly impact the cytosolic sorting motifs, a unique alteration of GNPTAB. Differences within the organization of the secretory pathway and the trafficking machinery within certain cell types may lead to differences in GlcNAc-1-phosphotransferase trafficking. We believe such differences may help explain the tissue-specific phenotypes seen within the GNPTAB c.10A>C/p.K4Q ML patients.
The reduced intracellular and much enhanced plasma activity of several acid glycosidases in this cohort does not differ significantly from the corresponding data obtained in the reference phenotypes, ML II and ML IIIαβ. The GNPT assays (Table 4) confirm earlier reports in leukocytes and fibroblasts that activity of the causative enzyme is nearly completely absent in ML II and between 2 and 20% of normal in ML IIIαβ.1, 8, 9, 16 The residual GNPT activity in each of the K4Q compound heterozygous ML lines is higher than that in ML II-derived fibroblasts and almost matches that in the ML IIIαβ line. Remarkably, but not unexpectedly, GNPT activity in the homozygous K4Q mutant cell line is 19.5% of that in controls and confirms the results previously obtained by others.17 This observation proves that the K4Q GNPTAB allele is indeed pathogenic but fairly leaky at least in cultured fibroblasts. The molecular characterization and trafficking of the enzyme may be further elucidated by additional studies of the mechanism by which this mutation alters GNPT activity.
The subnormal yet still effective maintenance of intracellular cathepsin D levels demonstrates the utility of this assay in distinguishing this specific intermediate type ML at least from the ML II reference phenotype. The fact that the majority of newly synthesized cathepsin D remains inside the cell in the GNPTAB c.10A>C/p.K4Q ML patients is surprising, and inconsistent with both the severity of the phenotype and the elevated plasma levels of lysosomal enzymes. However, these patient cells do retain substantial activity towards the simple sugar substrate, α-methylmannoside. We cannot rule the possibility that this mutation results in hydrolase-specific effects on lysosomal sorting. Further research on cathepsin-sorting efficiency may provide insight into the early failure of statural growth, nearly as severe as in ML II. The robust lysosomal enzyme missorting observed in ML II cells may extend to other cathepsins (for example, cathepsin K) known to play central roles in skeletal development. The relatively high efficiency of cathepsin D sorting may explain the moderate cognitive disability associated with this genotype. Roles for cathepsin D have been suggested in neural development and degeneration, and maintenance of near-normal intracellular levels of this protease may confer a protective effect within the brain of these patients.18, 19, 20
Delineation of this c.10A>C/p.K4Q GNPT-dependent intermediate ML has more than merely clinical and prognostic significance. The partial ‘uncoupling’ of physical and cognitive disability represents the apparently rare exception to the rule that nearly complete absence of GNPT activity yields the ML II phenotype and more residual activity results in ML IIIαβ.1 The severe DM, including the transient skeletal changes in infancy, is compatible with the ML II archetype. However, the specific intermediate type ML we describe is associated with GNPT activity and extension of life expectancy compatible with the MLIIIαβ archetype, and thus illustrates the progressively morbid clinical course not granted to the ML II patient. Manifestations include progressive and painful osteopenia, areas of frank osteolysis, and severe sclerosis in fasciae with contractures in tendons and other periarticular structures. Progressive destruction of the weight-bearing skeleton occurs both in the lower third of the ilia and particularly the femoral necks, occasionally resulting in pathological fracture. Clinical manifestations of osteoporosis are the late consequences of abnormal bone formation and excessive regional overmodeling, compatible with the observation of increased osteoclast recruitment to bone surfaces in some areas.21 Cyclic infusion of bisphosphonate may offer partial relief of pain as it is a potent analgesic in painful bone diseases and improves bone mineral density. The use of bisphosphonates is limited by the increasing risks for rare complications such as bisphosphonate-related atypical fractures. The gradual disappearance of the diaphyseal widening in the metacarpals in long-surviving patient 5 exemplifies the effects of late progressive overmodeling of qualitatively abnormal cortical and trabecular bone bone. The qualitative degrading and dysfunction of soft connective tissue adversely affects not only periarticular and peri-osseous structures, but also bronchopulmonary elasticity and cardiac valve anatomy and function, ultimately contributing to lethal outcome.2 Elucidation of the mechanisms by which this mutation leads to partial ‘uncoupling’ of the severe physical impact and the rather moderate cognitive disability in this specific intermediate ML will expand understanding of pathogenesis of ML beyond inadequately phosphorylated glycoproteins.
References
Cathey SS, Leroy JG, Wood T et al: Phenotype and genotype in mucolipidosis II and III alpha/beta: a study of 61 probands. J Med Genet 2010; 478–488.
Kobayashi H, Takahashi-Fujigasaki J, Takahiro F et al: Pathology of the first autopsy case diagnosed as mucolipidosis type III α/β suggesting autophagic dysfunction. Mol Genet Metab 2011; 102: 170–175.
Kozlowski K, Lipson A, Carey W : Mild I-cell disease, or severe pseudo-Hurler polydystrophy in three siblings; further evidence for intermediate forms of mucolipidosis II and III. Radiological features. Radiol Med 1991; 82: 847–851.
David-Vizcarra G, Briody J, Ault J et al: The natural history and osteodystrophy of mucolipidosis II and III. J Paediatr Child Health 2010; 46: 316–322.
Thomas GH, Taylor HA, Reynolds LW et al: Mucolipidosis 3 (Pseudo-Hurler polydystrophy): multiple lysosomal enzyme abnormalities in serum and cultured fibroblast cells. Pediatr Res 1973; 7: 751–756.
Braulke T, Geuze HJ, Slot JW et al: On the effects of weak bases and momensin on sorting and processing of lysosomal enzymes in human cells. Eur J Cell Biol 1987; 43: 316–321.
Steet R, Hullin R, Kudo M et al: A splicing mutation in the α/β GlcNAc-1-phosphotransferase gene results in an adult onset form of mucolipidosis III associated with sensory neuropathy and cardiomyopathy. Am J Med Genet A 2005; 132: 369–375.
Reitman ML, Kornfeld S : UDP-N-acetylglucosamine: glycoprotein N-acetylglucosamine-1-phosphtransferase proposed enzyme for the phosphorylation of the high mannose oligosaccharide units of lysosomal enzymes. J Biol Chem 1981; 256: 4275–4281.
Reitman ML, Kornfeld S : Lysosomal enzyme targeting. N-acetylglucosaminylphosphotransferase selectively phosphorylates native lysosomal enzymes. J Biol Chem 1981; 256: 11977–11980.
Spranger JW, Brill PW, Poznanski AK : Bone Dysplasias. An Atlas of Genetic Disorders of Skeletal Development 2nd (edn). New York: Oxford University Press, 2002, pp 261–262.
Pazzaglia UE, Beluffi G, Bianchi E et al: Study of bone pathology in early mucolipidosis II (I-Cell disease). Eur J Pediatr 1989; 148: 553–557.
Leroy JG, Cathey S, Friez MJ : Mucolipidosis II in: GeneReviews at Genetests: medical genetics information resource (database online). Copyright. Seattle: University of Washington, 2012, pp 1997-2010. Available at: http://www.genetests.org.
Leroy JG, Cathey S, Friez MJ : Mucolipidosis III alpha/beta in: GeneReviews at GeneTests: medical genetics information resource (database online). Copyright. Seattle: University of Washington, 2012, pp 1997-2010. Available at: http://www.genetests.org.
Reitman ML, Varki A, Kornfeld S : Fibroblasts from patients with I-cell disease and pseudo-Hurler polydystrophy are deficient in uridine 5’-diphosphate-N-acetylglucosamine: glycoprotein N-acetylglucosaminyl-phosphotransferase activity. J Clin Invest 1981; 67: 1574–1579.
Franke M, Braulke T, Storch S : Transport of the GlcNAc-1-phosphotransferase α/β-subunit precursor protein to the Golgi apparatus requires a combinatorial sorting motif. J Biol Chem 2012; 288: 1238–1249.
Varki AP, Reitman ML, Kornfeld S : Identification of a variant of mucolipidosis III (pseudo-Hurler polydystrophy): a catalytically active N-acetylglucosaminylphosphotransferase that fails to phosphorylate lysosomal enzymes. Proc Natl Acad Sci USA 1981; 78: 7773–7777.
Kudo M, Brem MS, Canfield WM : Mucolipidosis II (I-cell disease) and Mucolipidosis IIIA (classical pseudo-Hurler polydystrophy) are caused by mutations in the GlcNAc-phosphotransferase αβ subunits precursor gene. Am J Hum Genet 2006; 78: 451–463.
Amritraj A, Peake K, Kodam A et al: Increased activity and altered subcellular distribution of lysosomal enzymes determine neuronal vulnerability in Niemann-Pick type C1-deficient mice. Am J Pathol 2009; 175: 2540–2556.
Nakanishi H : Neuronal and microglial cathepsins in aging and age-related diseases. Ageing Res Rev 2003; 2: 367–381.
Tyynelä J, Sohar I, Sleat DE et al: A mutation in the ovine cathepsin D gene causes a congenital lysosomal storage disease with profound neurodegeneration. EMBO J 2000; 19: 2786–2792.
Robinson C, Baker N, Hoffman P et al: The osteodystrophy of mucolipidosis type III and the effects of intravenous pamidronate treatment. J Inherit Metab Dis 2002; 25: 681–693.
Acknowledgements
Funding has been provided by Award Number U54NS065768 from the National Institute of Neurological Disorders And Stroke and the Office of Rare Diseases Research (ORDR) through the Lysosomal Disease Network Consortium, a part of the NIH Rare Diseases Clinical Research Network (RDCRN), and by the National Institute of General Medical Sciences (GM086524). Additional support has been provided by ISMRD, the International Advocate for Glycoprotein Storage Diseases, the National MPS Society, and by the Genetics Endowment of South Carolina.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Leroy, J., Sillence, D., Wood, T. et al. A novel intermediate mucolipidosis II/IIIαβ caused by GNPTAB mutation in the cytosolic N-terminal domain. Eur J Hum Genet 22, 594–601 (2014). https://doi.org/10.1038/ejhg.2013.207
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.207
Keywords
This article is cited by
-
Early characteristic radiographic changes in mucolipidosis II
Pediatric Radiology (2016)
-
Association study of stuttering candidate genes GNPTAB, GNPTG and NAGPA with dyslexia in Chinese population
BMC Genetics (2015)
