
Abstract
Multiple sulfatase deficiency (MSD) is a rare inborn error of metabolism affecting posttranslational activation of sulfatases by the formylglycine generating enzyme (FGE). Due to mutations in the encoding SUMF1 gene, FGE’s catalytic capacity is impaired resulting in reduced cellular sulfatase activities. Both, FGE protein stability and residual activity determine disease severity and have previously been correlated with the clinical MSD phenotype. Here, we report a patient with a late infantile severe course of disease. The patient is compound heterozygous for two so far undescribed SUMF1 mutations, c.156delC (p.C52fsX57) and c.390A>T (p.E130D). In patient fibroblasts, mRNA of the frameshift allele is undetectable. In contrast, the allele encoding FGE-E130D is expressed. FGE-E130D correctly localizes to the endoplasmic reticulum and has a very high residual molecular activity in vitro (55% of wildtype FGE); however, it is rapidly degraded. Thus, despite substantial residual enzyme activity, protein instability determines disease severity, which highlights that potential MSD treatment approaches should target protein folding and stabilization mechanisms.
Similar content being viewed by others

Introduction
Multiple Sulfatase Deficiency (MSD, MIM: #272200) affects the post-translational activation of newly synthesized sulfatases by the formylglycine-generating enzyme (FGE).1 FGE is a glycoprotein of the endoplasmatic reticulum, 341 amino acids in length as mature enzyme, but is also secreted after N-terminal proteolytic processing.2 The crystal structure shows a monomeric fold with two buried calcium ions, cysteine pairs stabilizing the core domain and two catalytic cysteines directly involved in formylglycine-generating redox activity3, 4, 5 (Supplementary Figure S1). Mutations in the FGE-encoding SUMF1 gene lead to instable protein and/or impaired FGE catalytic function resulting in reduced sulfatase activities. More than 30 different SUMF1 mutations are known, most of them missense mutations that affect stability and residual molecular activity of mutant FGE, which both determine MSD disease severity.6, 7 The clinical phenotype includes the neonatal very severe, late infantile (severe and mild) and rare juvenile (mild) form. MSD combines main clinical symptoms of different single sulfatase deficiencies like dysmorphic features, neurodegenerative course of disease and ichthyotic skin rash. For patients with a functionally characterized mutation, a prediction of the clinical outcome is possible.6, 7, 8, 9 Here, we report a patient with two novel SUMF1 mutations, one nonsense and one missense, describe the clinical consequences and correlate this with the molecular defects attributable to the missense mutation.
Materials and methods
SUMF1 mutations and enzyme activity assays
Mutational analysis of the SUMF1 gene from genomic DNA of patient fibroblasts or leukocytes, as well as sulfatase and FGE activity assays were performed as described earlier.10, 11
Expression studies
Cell culturing of patient skin fibroblasts and HT-1080 fibrosarcoma cells as well as stable transfection with pSB-FGE-His plasmids were described earlier.6, 7 The FGE-E130D mutation was created by site-directed mutagenesis according to the QuikChange Protocol (Stratagene, La Jolla, CA, USA) with Pfu polymerase and complementary primer pairs (coding sequence only: FGE_E130D-His_c 5′-CATGGATGCCTATGATGTCAGTAATACTG-3′).
Western blot and immunofluorescence studies
Aliquots of 50-μg cell protein, from cells of a confluent T75 cell culture flask grown in 10 ml medium for 24 h, and 40 μl aliquots of this medium were analyzed by SDS–polyacrylamide gel electrophoresis and western blot using a polyclonal FGE antibody or monoclonal His antibody (Sigma Aldrich, Munich, Germany) as described.2 Endogenous FGE in skin fibroblasts or variant FGE in stably transfected HT-1080 cells was detected by indirect immunofluorescence using a polyclonal rabbit FGE antibody as described.2 Monoclonal antibodies as organelle markers were used against PDI (Biomol, Hamburg, Germany), LAMP1 (Abcam, Cambridge, UK) and Golgi protein 58K (Abcam, Cambridge, UK). Confocal images were taken on a Leica TCS SP2 AOBS laser-scanning microscope and analyzed with the manufacturer’s software (Leica, Solms, Germany).
FGE purification
HT-1080 cells stably overexpressing His-tagged wildtype or FGE-E130D were harvested, resuspended in buffer I (20 mM Tris-HCl, 40 mM imidazole, 500 mM sodium chloride, pH 8.0), and sonicated in presence of a protease inhibitor cocktail (Sigma Aldrich, Munich, Germany). The cell lysate was cleared by centrifugation at 100 000 × g for 20 min and loaded on a HisTrap column (GE Healthcare, Munich, Germany) using buffer I. The column was eluted in fractions of 1 ml with buffer II (20 mM Tris-HCl, 500 mM sodium chloride 500 mM imidazole, pH 8.0) running a gradient from 0–100% of buffer II in 25 min on a ÄKTA system (GE Healthcare). Appropriate elution fractions were pooled (6–8 ml), and concentrated to 50 μl using Amicon centrifugation filter units (Millipore, Billerica, MA, USA). The concentrated eluate was dialyzed against buffer III (20 mM Tris-HCl, pH 8), its FGE amount was determined and used for FGE activity assays.
Metabolic labeling and immunoprecipitation
HT-1080 cells stably expressing FGE-His or FGE-E130D-His were subjected to metabolic labeling with 35S-methionine/cysteine and immunoprecipitation followed by phosphorimaging and densitometric quantification (PMI System, Biorad, Munich, Germany) as described.7
Results
A patient with MSD displays a late infantile severe phenotype and reduced sulfatase activities
The female patient was born as the second child of non-consanguineous parents from Switzerland. In the first months she presented with muscular hypotonia and had a consecutive developmental delay. She was able to sit at age 14 months; at age 20 months she could grasp, babble, and was visually interactive. At age 22 months progressive leg spasticity became obvious, followed by paroxysmal tonic upward eye movements at age 25 months. At this time she started to lose motor and visual skills. Cerebral magnetic resonance imaging at age 27 months displayed a leukoencephalopathy pattern as seen in metachromatic leukodystrophy. Laboratory examinations revealed high-urine concentrations of glycosaminoglycans as well as clearly reduced activity of three cellular sulfatases tested (Table 1). At age 31 months she showed signs of tetraplegia, long eye lashes, a dry and scaly skin and no more reactions to noises. At 4 years and 1 month, coarse facies and dysostosis multiplex were detected, organomegaly was still absent. From the age of 39 months on she had irregular breathing attacks due to intermittent central hypoventilation. She died at the age of 5 years and 5 months (for a clinical synopsis see Table 2). The course of the disease in this patient could be classified as late infantile severe form of MSD. The early onset of neurodegeneration in the second year of life and the disease progression due to the manifestation of several additional clinical symptoms throughout her life is comparable to other described late infantile severe cases of MSD.6, 7, 8
Two new MSD causing SUMF1 mutations
In this patient we identified two so far unknown SUMF1 mutations. c.156delC leads to a frameshift and premature stop codon (p.C52fsX57). On the other allele, the single nucleotide exchange c.390A>T substitutes glutamate at position 130 of FGE by aspartate. At the mRNA level only the nucleotide exchange c.390A>T was detectable (data not shown).
FGE-E130D: correct intracellular localization, exceptionally high residual activity but drastically impaired intracellular stability and no secretion
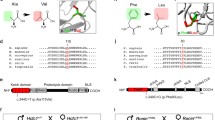
Endogenous FGE-E130D in patient fibroblasts was hardly detectable (Supplementary Figure S2). For detailed analysis we expressed recombinant His-tagged FGE-E130D in human HT-1080 fibrosarcoma cells. FGE-E130D was correctly localized in the endoplasmatic reticulum (Figure 1a). Western blot analysis (Figure 1b) revealed that FGE-E130D was not secreted, whereas a major fraction of wildtype FGE was secreted in a N-terminally truncated form (38 kDa, starting at glutamate-73) due to proteolytic processing during secretion, as described previously.2 Pulse-chase analysis by metabolic 35S-labeling indicated a severe reduction of the intracellular stability of FGE-E130D, such that about 90% of the protein was degraded within 3 h (Figure 1d). Partially purified FGE-E130D displayed a residual FGE activity of 55 +/−16% (Figure 1c). Of note, the intracellular FGE-E130D was present in two molecular forms, one with a molecular mass of 41 kDa, comparable to wildtype FGE, and the other with 43–44 kDa. Endoglycosidase H treatment converted both forms to a 39-kDa form (data not shown).

Localization, expression, activity and stability of FGE-E130D in HT-1080 cells. (a) Immunofluorescence detection of FGE (red) in stably transfected cells indicates that FGE-E130D, like wildtype FGE, co-localizes with the endoplasmatic reticulum marker protein disulfide isomerase (PDI, green), as shown in the merge. Untransfected cells (control) show endogenous FGE expression. (b) Western blot analysis of recombinant FGE-E130D, in comparison to wildtype FGE. Equal amounts of total cell lysate and medium were analyzed. Due to instability (see panel D) FGE-E130D is expressed at markedly reduced steady-state levels. The asterisk labels a specific band observed for FGE-E130D that is sensitive to glycosidase treatment (see text). (c) Residual activity of FGE-E130D was measured after partial purification of the protein from transfected cells. Defined amounts of FGE protein (27 ng, upper panel) were used for the assay. Measured activity is given relative to that of wildtype FGE, which was purified and incubated under identical conditions in parallel (mean values +/− SD, n=3). (d) Stability of FGE-E130D. After metabolic labeling, FGE protein was immunoprecipitated from cells and medium either immediately, 1.5 or 3 h after labeling. The intensity of bands was quantified and expressed as percentage of the protein amount present at 0 h (see lower panel). Note that about equal amounts of wildtype and mutant FGE were present at 0 h; however, FGE-E130D vanished almost completely from the cells within three hours and was not secreted.
Discussion
MSD is still an untreatable disease. Thus, potential treatment approaches, and also genetic counseling, directly depend on thorough analyses of the functional consequences of human SUMF1 mutations on the clinical and biochemical phenotype. In a severely affected MSD patient with the late infantile onset of disease we identified and characterized two yet undescribed SUMF1 mutations, namely c.156delC and c.390A>T. As no corresponding mRNA for the single nucleotide deletion (p.C52fsX57) could be detected, probably in consequence of nonsense mediated decay,12 the resulting sulfatase acitivities and apparently also the clinical phenotype are due to the missense mutation c.390A>T leading to expression of FGE-E130D protein.
The E130D mutation severely destabilizes FGE
Missense mutations represent the majority of genetic defects in MSD. For eight different missense mutations the instability of mutant FGE, as predicted from the crystal structure, could be verified by stability and residual activity measurements.4, 6, 7 In FGE-E130D the aspartate-130 side-chain is too short to position one of two calcium ions in the coordination plane. Consequently, interactions with adjacent residues are disturbed (Supplementary Figure S1) leading to protein destabilization and most likely early degradation.13 Indeed, we could show that FGE-E130D is unable to pass endoplasmatic reticulum quality control, hence getting rapidly and completely degraded inside cells without entering into secretion. Interestingly, we found expression of FGE-E130D with two different molecular weights in all experiments and even after purification (Figures 1b and d). Both forms were sensitive to treatment with endoglycosidase H leading to a deglycosylated 39-kDa protein (data not shown). Details of these two alternative glycosylation patterns remain to be elucidated.
FGE-E130D displays very high residual catalytic activity
Up to now, all structurally impaired FGE variants showed clearly reduced specific enzyme activity.6, 7 The residual catalytic activities have been determined with purified FGE variants and reflect the true molecular enzymatic capacity of the mutant proteins in vitro. FGE-E130D shows a remarkable residual activity (55% compared with wildtype). So far, this is the highest residual activity that has been reported for a mutant FGE found in MSD. Such a high activity, if true in vivo, would be more than enough to maintain full sulfatase activation. However, the patient’s compromised activities of sulfatases are in the range of no more than 10–20% of control activities, hence indicating massive FGE deficiency. This obviously is a consequence of the rapid degradation of FGE-E130D. Thus, protein instability is the leading molecular factor for pathophysiology in the reported patient.
Genotype-phenotype relationship in severe late infantile MSD
The clinical course of MSD in the reported patient corresponds to the late infantile severe form. Generally, MSD patients with high residual FGE activity, but instable protein reveal an attenuated course of disease.6, 7 In contrast to FGE-E130D, as reported here, all other mutant FGE proteins with high residual activity in former studies were found to be secreted. This indicates that these mutants at least in part were successful to pass intracellular control mechanisms of protein folding. The specific biochemical feature of a rapidly degraded (and hence non-secreted) FGE-E130D almost certainly contributes to the late infantile severe phenotype of our patient, thereby highlighting the importance of affected FGE stability and protein degradation as a main pathophysiological cause for MSD and as a reliable indicator of disease severity in case of high residual FGE activity. This does not exclude that other so far undiscovered disease modifiers also influence the clinical phenotypes of MSD patients. Our patient report underlines that potential future therapies should aim at protein stabilization mechanisms −in particular when residual mutant FGE activities are high.
References
Dierks T, Schlotawa L, Frese MA, Radhakrishnan K, von Figura K, Schmidt B : Molecular basis of multiple sulfatase deficiency, mucolipidosis II/III and Niemann-Pick C1 disease - Lysosomal storage disorders caused by defects of non-lysosomal proteins. Biochim Biophys Acta 2009; 1793: 710–725.
Preusser-Kunze A, Mariappan M, Schmidt B et al: Molecular characterization of the human Calpha-formylglycine-generating enzyme. J Biol Chem 2005; 280: 14900–14910.
Roeser D, Preusser-Kunze A, Schmidt B et al: A general binding mechanism for all human sulfatases by the formylglycine-generating enzyme. Proc Natl Acad Sci USA 2006; 103: 81–86.
Dierks T, Dickmanns A, Preusser-Kunze A et al: Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell 2005; 121: 541–552.
Mariappan M, Gande SL, Radhakrishnan K, Schmidt B, Dierks T, von Figura K : The non-catalytic N-terminal extension of formylglycine-generating enzyme is required for its biological activity and retention in the endoplasmic reticulum. J Biol Chem 2008; 283: 11556–11564.
Schlotawa L, Steinfeld R, von Figura K, Dierks T, Gartner J : Molecular analysis of SUMF1 mutations: stability and residual activity of mutant formylglycine-generating enzyme determine disease severity in multiple sulfatase deficiency. Human Mutat 2008; 29: 205.
Schlotawa L, Ennemann EC, Radhakrishnan K et al: SUMF1 mutations affecting stability and activity of formylglycine generating enzyme predict clinical outcome in multiple sulfatase deficiency. Eur J Hum Genet 2011; 19: 253–261.
Eto Y, Gomibuchi I, Umezawa F, Tsuda T : Pathochemistry, pathogenesis and enzyme replacement in multiple-sulfatase deficiency. Enzyme 1987; 38: 273–279.
Blanco-Aguirre ME, Kofman-Alfaro SH, Rivera-Vega MR et al: Unusual clinical presentation in two cases of multiple sulfatase deficiency. Pediatr Dermatol 2001; 18: 388–392.
Dierks T, Schmidt B, Borissenko LV et al: Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell 2003; 113: 435–444.
Rommerskirch W, von Figura K : Multiple sulfatase deficiency: catalytically inactive sulfatases are expressed from retrovirally introduced sulfatase cDNAs. Proc Natl Acad Sci USA 1992; 89: 2561–2565.
Amrani N, Sachs MS, Jacobson A : Early nonsense: mRNA decay solves a translational problem. Nat Rev Mol Cell Biol 2006; 7: 415–425.
Maattanen P, Gehring K, Bergeron JJ, Thomas DY : Protein quality control in the ER: the recognition of misfolded proteins. Semin Cell Dev Biol 2010; 21: 500–511.
Acknowledgements
We thank Nicole Hollstein, Nicole Eiselt, Tanja Wilke, Hendrik Rosewich, Eva Ennemann, Michaela Wachs, Md Sarfaraz Alam and Achim Dickmanns for technical assistance and discussions. Special thanks go to Kurt von Figura for implementing the project and Peter Rehling for continuous support. This work was supported by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Schlotawa, L., Radhakrishnan, K., Baumgartner, M. et al. Rapid degradation of an active formylglycine generating enzyme variant leads to a late infantile severe form of multiple sulfatase deficiency. Eur J Hum Genet 21, 1020–1023 (2013). https://doi.org/10.1038/ejhg.2012.291
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.291
Keywords
This article is cited by
-
Late infantile form of multiple sulfatase deficiency with a novel missense variant in the SUMF1 gene: case report and review
BMC Pediatrics (2023)
-
Types and effects of protein variations
Human Genetics (2015)
-
Multiple sulfatase deficiency with neonatal manifestation
Italian Journal of Pediatrics (2014)
-
A non-conserved miRNA regulates lysosomal function and impacts on a human lysosomal storage disorder
Nature Communications (2014)
