
Abstract
Wolfram syndrome (WFS) is a neurodegenerative genetic condition characterized by juvenile-onset of diabetes mellitus and optic atrophy. We studied clinical features and the molecular basis of severe WFS (neurodegenerative complications) in two consanguineous families from Iran. A clinical and molecular genetic investigation was performed in the affected and healthy members of two families. The clinical diagnosis of WFS was confirmed by the existence of diabetes mellitus and optic atrophy in the affected patients, who in addition had severe neurodegenerative complications. Sequencing of WFS1 was undertaken in one affected member from each family. Targeted mutations were tested in all members of relevant families. Patients had most of the reported features of WFS. Two affected males in the first family had fathered unaffected children. We identified two homozygous mutations previously reported with apparently milder phenotypes: family 1: c.631G>A (p.Asp211Asn) in exon 5, and family 2: c.1456C>T (p.Gln486*) in exon 8. Heterozygous carriers were unaffected. This is the first report of male Wolfram patients who have successfully fathered children. Surprisingly, they also had almost all the complications associated with WFS. Our report has implications for genetic counseling and family planning advice for other affected families.
Similar content being viewed by others

Introduction
Wolfram syndrome (WFS), first described by Wolfram and Wagener in 1938, is a severe genetic neurodegenerative disease with autosomal recessive inheritance. WFS is mainly characterized by juvenile-onset diabetes mellitus and optic atrophy.1, 2 The other features of this syndrome include diabetes insipidus, sensorineural hearing loss, peripheral neuropathy, ataxia, psychiatric problems, renal-tract abnormalities, bladder atony and male hypogonadism.3, 4, 5
The exact world-wide prevalence of WFS is unknown but it has been estimated to be 1 per 770 000 in the UK, with a carrier frequency of 1 in 354.3 The prevalence in North American population was estimated at 1 per 100 000.6
The most common condition in WFS is diabetes mellitus that starts in the first decade of life. The main causes of morbidity and mortality are complications of neurodegeneration and urinary tract infections.7
There is no treatment available for WFS. The mortality is high and only about 40% of patients survive beyond the age of 35 years.7 The cause of death is usually central respiratory failure due to brainstem atrophy in the third or fourth decade.3
WFS1 is the main gene identified as the cause of WFS.8, 9 This gene is located on chromosome 4p16.3 and encodes Wolframin, a transmembrane protein that maintains calcium homeostasis of the endoplasmic reticulum (ER).8, 9, 10, 11 Other roles, such as regulation of ER stress and protein biosynthesis, modification/ folding, have also been suggested for Wolframin.12, 13, 14, 15 Wolframin is an 890 amino acid oligomeric protein with a molecular mass of 100 kDa.8, 9
Although many mutations have been identified in the WFS1 gene, most affected patients carry a mutation, either insertion, deletion, nonsense or missense, in exon 8, the largest exon of WFS1.8, 9, 16, 17, 18, 19 The C-terminal hydrophilic part of the protein is the major site for missense mutations. Studies indicate that mutations affecting the translation of the last 10–15 amino acids result in a severe disease phenotype that confirms the functional importance of the C-terminus.17, 20
The exact function of WFS1 Wolframin protein is still unknown. There is also no clear genotype–phenotype correlation. There are several case reports of affected females having unaffected children; but no reports of male patients having successfully fathered children. Data from mouse knockout models show reduced spermatogenesis and reduced fertility in males compared with females.21
We report two affected families from Iran with a severe phenotype, in one of which affected males were able to father children.
Materials and methods
Patients and families
We studied six patients from two Persian families. The families were consanguineous and the minimum diagnostic criteria for WFS were childhood occurrence of diabetes mellitus and optic atrophy.3 Written consent to participate in the study was obtained from members of both families.
All members of both families underwent comprehensive physical exams, including endocrinological, ophthalmological, neurological, urogenital and psychiatric assessments.
Methods
Blood samples were collected from each subject and genomic DNA was extracted from peripheral leukocytes using QIAamp DNA blood Midi kits (Qiagen, Hilden, Germany).
Direct sequencing of all the exons and intron–exon boundaries of WFS1 was performed on genomic DNA of the probands from two families.
The WFS1 gene coding region was analyzed by PCR amplification of genomic DNA using primers previously described.22 The PCR products were directly sequenced on both strands using Big Dye v.3.1 chemistry on ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA) and sequences were compared with human genomic and cDNA WFS1 sequences (GenBank accession no. AF084481).
Segregation analysis was performed in other patients and healthy members of the families. This was done by sequencing for the mutation and carrier status in relatives of the affecteds.
Paternity testing was undertaken for each father/child pair, genotyping both alleles for 15 independent loci across the genome (Micropathology Ltd, Coventry, UK).
Results
Clinical findings
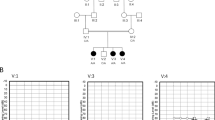
The multi-generation family (Figure 1a) included three WFS patients; two males and one female. The parents were first cousins and healthy. The proband (family 1:IV-2) was a 35-year-old male with short stature. He was married with a healthy 4-year-old son (family 1:V-1). His symptoms started from the age of 7 with polyuria and polydipsia and high blood glucose (335 mg/dl). He was diagnosed with diabetes mellitus but his polyuria continued despite reasonable glycemic control. During an ophthalmological check at age 10, bilateral optic atrophy was detected. In a recent ophthalmological assessment, both eyes were almost blind (visual acuity 6/60 or less). His hearing ability decreased in the late third decade of life. Neurological examination revealed hyperreflexia in the lower limbs. Ultrasonography demonstrated hydronephrosis, hydroureter and increased post-void residual urine. Blood laboratory findings included blood urea: 32 g/dl, Creatinine: 1.5 g/dl and HbA1c 7.8%. Urinalysis revealed specific gravity 1016 and glucose 2+. Full blood count and electrolytes were in normal range. His affected brother (family 1:IV-5, 31 years) and sister (family 1:IV-8, 25 years) had similar patterns of disease onset and features, but did not have any hearing problem. Her mean HbA1C level was 8.7 and the other laboratory data were otherwise normal. The affected brother had a healthy 3-year-old son and his laboratory findings were completely normal with a mean HbA1C of 8.3. One of their male cousins (family 1:IV-10) had died from WFS a few years earlier. In the second family (Figure 1b) consisting of five alive siblings (four female, one male) and one deceased female sibling, two sisters and the brother were affected by WFS. Their parents were first cousins once removed and without any known health problem. The proband (family 2:II-5, 23 years, male) was diagnosed with diabetes mellitus at the age of 9, with polyuria, polydipsia and hyperglycemia (355 mg/dl). Polyuria was not controlled after insulin therapy for diabetes. He was diagnosed with diabetes insipidus (urine specific gravity 1008) at age 15 years. The patient had primary enuresis, together with bilateral hydronephrosis and hydroureter on ultrasonography, thought to be due to neurogenic bladder. He developed renal failure at 17 years of age requiring hemodialysis. Ophthalmological examination at age 8 years had revealed optic atrophy and a recent eye test showed visual acuity <6/60 in both eyes. His hearing difficulties had started at the age of 12 years and resulted in sensorineural deafness by the late second decade of life. He also had short stature. On neurodevelopmental evaluation, the patient had cerebellar ataxia, poor concentration and moderate intellectual disability. Neurological assessment demonstrated hyporreflexia in lower limbs, whereas upper limbs were normal. He complained of epigastric pain that had started 1 year earlier. The patient had vesico-ureteric reflux on voiding cystogram, and urinary retention necessitating intermittent catheterization. Laboratory tests revealed the following results; Hb: 8.75 g/dl, urea: 65 g/dl, Creatinine: 2.5 g/dl, HbA1c(%): 10.4 and PTH: 118 pg/ml (normal <69). Urinalysis findings included specific gravity: 1007, pH: 5.0, glucose 2+, protein 1+ and WBC 35–40/hpf, electrolytes were normal. Other affected siblings of this family also had features of diabetes mellitus, optic atrophy, deafness, diabetes insipidus, neurogenic bladder and psychiatric problems with similar pattern of onset. The laboratory data of (family 2:II-2) and (family 2:II-6) was within normal limits with the mean HbA1C level of 9.6 and 8.8, respectively. Furthermore, the deceased sibling, who died at the age of 30 because of renal failure, had proximal muscle weakness, and electromyography (EMG) and nerve conduction velocity (NCV) investigations had revealed axonal involvement in the peripheral nerves.

Pedigrees of the WFS families. (a) Family 1 and (b) family 2. Affected individuals are indicated with filled symbols. wt: wild-type allele; mut: mutant allele (c.631G >A in family 1 and c.1456C >T in family 2).
Thyroid function tests, lipid profiles and hepatic function were all normal, and diabetes autoimmune markers were negative in all patients from both families.
Molecular study
Molecular analysis of the WFS1 gene in the patients from the first family identified that all were homozygous for a missense mutation in exon 5, c.631G>A that alters aspartic acid to asparagine at amino acid position 211.
In the patients from the second family, sequencing of their genomic DNAs revealed homozygosity for a nonsense mutation, c.1456C>T that substitutes a glutamine at position 486 for a stop codon, predicted to cause truncation of wolframin. This mutation is located in exon 8 of WFS1.
Parents and healthy siblings in both families were heterozygous for the relevant mutation.
All members (affected and healthy) of two families were homozygous for a non-synonymous polymorphism, c.997A>G (p.Ile333Val, rs1801212). Moreover, all members of the second family were homozygous for another non-synonymous polymorphism, c.1832G>A (p.Arg611His, rs734312). Both SNPs are located in exon 8 of WFS1.
The two affected male members of the first family and their two unaffected children underwent paternity testing by genotyping for 15 markers spread across the genome. For each of these 15 independent loci, the fathers possessed alleles that were present in their children. The probability that the samples originated from the true biological fathers of the children compared with any other male from a random population is >99.9%.
Discussion
WFS in Iran was first described in two siblings.23 The probands presented with diabetes mellitus (at the age of 6 years) followed by bilateral optic atrophy in the first decade. The other associated features included diabetes insipidus and sensorineural deafness (in the second decade), dilated renal outflow tracts (early in the third decade) and multiple neurological abnormalities (early in the fourth decade of life).23 In our study, two families of Persian origin presented with a severe form of WFS exhibiting almost all of the reported features.
The c.631G>A (p.Asp211Asn) mutation was first described by Van ven Ouweland et al18 in a heterozygous state in two affected sisters (patients 1 and 2, Table 1). They described a non-consanguineous German family with three affected siblings (two alive daughters and one deceased son) suffering from relatively mild phenotype and relatively late onset of optic atrophy and the absence of renal tract or neurological abnormalities. The patients had developed diabetes mellitus and optic atrophy in early second (10, 12 years) and late third decade of life (28, 26 years), respectively. The associated conditions included deafness in one patient, and hypothyroidism and hypertension in the other. Both sisters were compound heterozygotes for two missense mutations; c.631G>A (p.Asp211Asn) and c.1820C>G (p.Pro607Arg).18 The latter was found in one of their healthy brothers as well. Table 1 compares the clinical features of these sisters and patients from family 1. Considering the mild clinical features of two other missense mutations in the literature (A716T24 and P885L17), Van ven Ouweland et al18 suggested a correlation between the type of the mutation and severity of the disease; homozygosity or compound heterozygosity for missense mutations lead to an attenuated form of the disease, whereas protein-truncating mutations may cause severe WFS. This is in contrast to our findings that describe the same missense mutation associated with severe WFS, defined as neurodegenerative involvement.
The c.631G>A is located on exon 5 of WFS1. To date, seven causative mutations have been identified within exon 5, of which five mutations17, 18, 19, 25, 26 result in WFS, whereas two mutations27, 28 are associated with low-frequency sensorineural hearing loss. p.Asp211Asn is located in the N-terminal domain of the WFS1 protein. This mutation was not associated with low-frequency hearing impairment in the heterozygous carriers.18, 29 ClustalW analysis30 of WFS1 revealed this aspartic acid residue is conserved in different mammals (Figure 2).

Multiple sequence alignment of the WFS1 protein region flanking residues Asp211 and Gln486. ClustalW analysis demonstrates that amino acids, aspartic acid at position 211 and glutamine at position 486, are well conserved in orthologs. Missense mutations are indicated by arrows.
The c.1456C >T (p.Gln486*) was identified in a mother and daughter from a non-consanguineous Italian family. They were both heterozygous for this nonsense mutation that is located in exon 8 of the WFS1 gene. The proband (patient 3, Table 1) had developed diabetes mellitus and optic atrophy at the age of 4 and 13 years, respectively. The additional clinical conditions included diabetes insipidus (onset age of 9 years), deafness (7 years) and urinary tract dilation.16 Table 1 compares the clinical features of this patient and patients from family 2. The c.1456C>T truncates the WFS1 protein after the fifth transmembrane segment resulting in a non-functional protein.31 The p.Gln486* is located in the EC3 domain of WFS1 protein and conserved in different mammals (Figure 2).
Both mutations (p.Asp211Asn and p.Gln486*) we identified in this study were disease-causing only in their homozygous status and heterozygous individuals did not demonstrate any feature of WFS (Figure 1).
As reported above, two male patients in the first family had children. To the best of our knowledge, this is the first report of two male patients (family 1:IV-2 and IV-5) with most of the complications of WFS fathering children (family 1:V-1 and V-2), who were unaffected, and heterozygous carriers for the relevant identified mutations (Figure 1a) and polymorphisms. Paternity testing using 15 independent loci from across the genome showed that paternity was 99.9% certain compared with another random male from the same population. Clearly there is a possibility that a related male within the family fathered the children; this possibility cannot be ruled out.
Male hypogonadism has been a well reported feature of WFS,3, 32, 33, 34, 35, 36, 37, 38 in contrast to affected females, for whom there are several reports of successful pregnancies in the literature.21 Gonadal dysfunction in WFS male and female patients was first reported in 1977.4 Peden et al39 also reported three male WFS patients, all with gonadal abnormalities (primary hypogonadism, atrophic testes and delayed maturation). One patient was a 31-year-old male with the gonadal features of small penis and small soft testes although with normal facial, axillary and pubic hair. Hormone studies revealed basal serum testosterone 6.8 nmol/1 (normal 10–30), basal LH 40 mU/1 and basal FSH 31 mU/1. The patient had never had penile erection or ejaculation.39
In another study by Barrett et al,3 7 of 10 WFS males had primary gonadal atrophy (follicle stimulating hormone >20 μ/l, luteinising hormone >16 jjb/l, testosterone <12 nmol/l). Given the appropriate level of gonadotropic hormones (basal elevations of LH and FSH and exaggerated responsiveness to LHRH) and normal growth of the patients, researchers concluded that the patients probably had primary hypogonadism.3, 39
Noormets et al21 used a mouse model to investigate the role of WFS1 gene in infertility. They generated WFS1-deficient (WFS1KO) male mice using homologous recombination. The results showed impairment of the fertility in WFS1KO males. The motility and morphology of the sperm and the histology of testes were analyzed. In the WFS1KO males, sperm motility was not affected but their spermatozoa had abnormal morphology compared with wild-type males. Testis histology of the WFS1KO males also demonstrated significantly reduced number of spermatogonia and sertoli cells. WFS1KO males had normal concentration of serum testosterone and FSH concentrations.21 The Wolframin protein is likely to have several functions, being involved in calcium homeostasis and possibly other ion transport, as well as a negative regulator of ER stress. It is possible that disruption of ion homeostatis affects sertoli cell development and sperm maturation/function, leading to reduced fertility.
The combination of severe phenotype in our families associated with fertility in males is paradoxical. One possible explanation is that our affected family members are older than other reports in the literature, so have had time for symptoms to progress.
In summary, we report severe phenotypes in families affected by previously reported mutations associated with mild phenotypes. We also report the first examples of male fertility. Our findings have implications for genetic counseling for future families affected by these mutations, and raise the hope for that at least some affected males may be able to father children.
Accession codes
References
Wolfram DJ, Wagener HP : Diabetes mellitus and simple optic atrophy among siblings: report of four cases. Mayo Clin Proc 1938; 13: 715–718.
Blasi C, Pierelli F, Rispoli E, Saponara M, Vingolo E, Andreani D : Wolfram's syndrome: a clinical, diagnostic, and interpretative contribution. Diabetes Care 1986; 9: 521–528.
Barrett TG, Bundey SE, Macleod AF : Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995; 346: 1458–1463.
Cremers CW, Wijdeveld PG, Pinckers AJ : Juvenile diabetes mellitus, optic atrophy, hearing loss, diabetes insipidus, atonia of the urinary tract and bladder, and other abnormalities (Wolfram syndrome). A review of 88 cases from the literature with personal observations on 3 new patients. Acta Paediatr Scand Suppl 1977; 264: 1–16.
Minton JA, Rainbow LA, Ricketts C, Barrett TG : Wolfram syndrome. Rev Endocr Metab Disord 2003; 4: 53–59.
Fraser FC, Gunn T : Diabetes mellitus, diabetes insipidus, and optic atrophy. An autosomal recessive syndrome? J Med Genet 1977; 14: 190–193.
Kinsley BT, Swift M, Dumont RH, Swift RG : Morbidity and mortality in the Wolfram syndrome. Diabetes Care 1995; 18: 1566–1570.
Inoue H, Tanizawa Y, Wasson J et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet 1998; 20: 143–148.
Strom TM, Hortnagel K, Hofmann S et al. Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum Mol Genet 1998; 7: 2021–2028.
Takeda K, Inoue H, Tanizawa Y et al. WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet 2001; 10: 477–484.
Osman AA, Saito M, Makepeace C, Permutt MA, Schlesinger P, Mueckler M : Wolframin expression induces novel ion channel activity in endoplasmic reticulum membranes and increases intracellular calcium. J Biol Chem 2003; 278: 52755–52762.
Zatyka M, Ricketts C, da Silva Xavier G et al. Sodium-potassium ATPase 1 subunit is a molecular partner of Wolframin, an endoplasmic reticulum protein involved in ER stress. Hum Mol Genet 2008; 17: 190–200.
Yamaguchi S, Ishihara H, Tamura A et al. Endoplasmic reticulum stress and N-glycosylation modulate expression of WFS1 protein. Biochem Biophys Res Commun 2004; 325: 250–256.
Hofmann S, Bauer MF : Wolfram syndrome-associated mutations lead to instability and proteasomal degradation of wolframin. FEBS Lett 2006; 580: 4000–4004.
Yurimoto S, Hatano N, Tsuchiya M et al. Identification and characterization of wolframin, the product of the wolfram syndrome gene (WFS1), as a novel calmodulin-binding protein. Biochemistry 2009; 48: 3946–3955.
Colosimo A, Guida V, Rigoli L et al. Molecular detection of novel WFS1 mutations in patients with Wolfram syndrome by a DHPLC-based assay. Human mutation 2003; 21: 622–629.
Hardy C, Khanim F, Torres R et al. Clinical and molecular genetic analysis of 19 Wolfram syndrome kindreds demonstrating a wide spectrum of mutations in WFS1. Am J Hum Genet 1999; 65: 1279–1290.
van ven Ouweland JM, Cryns K, Pennings RJ et al. Molecular characterization of WFS1 in patients with Wolfram syndrome. J Mol Diagn 2003; 5: 88–95.
Gasparin MR, Crispim F, Paula SL et al. Identification of novel mutations of the WFS1 gene in Brazilian patients with Wolfram syndrome. Eur J Endocrinol 2009; 160: 309–316.
Sam W, Qin H, Crawford B, Yue D, Yu S : Homozygosity for a 4-bp deletion in a patient with Wolfram syndrome suggesting possible phenotype and genotype correlation. Clin Genet 2001; 59: 136–138.
Noormets K, Koks S, Kavak A et al. Male mice with deleted Wolframin (Wfs1) gene have reduced fertility. Reprod Biol Endocrinol 2009; 7: 82.
Minton JA, Hattersley AT, Owen K et al. Association studies of genetic variation in the WFS1 gene and type 2 diabetes in U.K. populations. Diabetes 2002; 51: 1287–1290.
Manaviat MR, Rashidi M, Mohammadi SM : Wolfram Syndrome presenting with optic atrophy and diabetes mellitus: two case reports. Cases J 2009; 2: 9355.
Young TL, Ives E, Lynch E et al. Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum Mol Genet 2001; 10: 2509–2514.
Zenteno JC, Ruiz G, Perez-Cano HJ, Camargo M : Familial Wolfram syndrome due to compound heterozygosity for two novel WFS1 mutations. Mol Vis 2008; 14: 1353–1357.
Hansen L, Eiberg H, Barrett T et al. Mutation analysis of the WFS1 gene in seven Danish Wolfram syndrome families; four new mutations identified. Eur J Hum Genet 2005; 13: 1275–1284.
Tranebjaerg L, Hansen L, Bille M et al. Low frequency hearing impairment due to GJB2 and WFS1 mutations shows unrecognized genetic heterogeneity. Presented at the Molecular Biology of Hearing and Deafness meeting 2004 Bethesda, MD, USA.
Cryns K, Sivakumaran TA, Van den Ouweland JM et al. Mutational spectrum of the WFS1 gene in Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus, and psychiatric disease. Hum Mutat 2003; 22: 275–287.
Pennings RJ, Huygen PL, van den Ouweland JM et al. Sex-related hearing impairment in Wolfram syndrome patients identified by inactivating WFS1 mutations. Audiol Neurootol 2004; 9: 51–62.
Thompson JD, Higgins DG, Gibson TJ : CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994; 22: 4673–4680.
Alimadadi A, Ebrahim-Habibi A, Abbasi F, Amoli M, Sayahpour FA, Larijani B : Novel mutations of wolframin: a report with a look at the protein structure. Clin Genet 2011; 79: 96–99.
Homan MR, MacKay BR : Primary hypogonadism in two siblings with Wolfram syndrome. Diabetes Care 1987; 10: 664–665.
Medlej R, Wasson J, Baz P et al. Diabetes mellitus and optic atrophy: a study of Wolfram syndrome in the Lebanese population. J Clin Endocrinol Metab 2004; 89: 1656–1661.
Cooper IS, Rynearson EH, Bailey AA, Mac CC : The relation of spinal cord diseases to gynecomastia and testicular atrophy. Proc Staff Meet Mayo Clin 1950; 25: 320–326.
Barjon P, Labauge R, Cazaban R, Fabre S : Primary Optic Atrophy and Neurogenic Deafness in 2 Diabetic Brothers. Diabete 1963; 11: 331–337.
Dedes W, Wildberger H, Landau K : Optic atrophy as a sign of wolfram syndrome. Klin Monbl Augenheilkd 2005; 222: 248–251.
Simsek E, Simsek T, Tekgul S, Hosal S, Seyrantepe V, Aktan G : Wolfram (DIDMOAD) syndrome: a multidisciplinary clinical study in nine Turkish patients and review of the literature. Acta Paediatr 2003; 92: 55–61.
Fernandez Rodriguez A, Gomez Balaguer M, Santolaya Garcia JI, Canto Faubel E, Carbonell Ferrer JM, Polo Peris : Uro-andrologic alterations in Wolfram syndrome. Arch Esp Urol 1991; 44: 871–873.
Peden NR, Gay JD, Jung RT, Kuwayti K : Wolfram (DIDMOAD) syndrome: a complex long-term problem in management. Q J Med 1986; 58: 167–180.
Acknowledgements
We thank Marziye Mohamadi Anaie for assistance with DNA extraction, and Dr Phil Bennett and Micropathology Ltd for paternity testing. This study was undertaken as part of GENE-ME (a research initiative for investigation of inherited diseases from the Middle-East).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Haghighi, A., Haghighi, A., Setoodeh, A. et al. Identification of homozygous WFS1 mutations (p.Asp211Asn, p.Gln486*) causing severe Wolfram syndrome and first report of male fertility. Eur J Hum Genet 21, 347–351 (2013). https://doi.org/10.1038/ejhg.2012.154
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.154
Keywords
This article is cited by
-
A mutant wfs1 zebrafish model of Wolfram syndrome manifesting visual dysfunction and developmental delay
Scientific Reports (2021)
-
Molecular investigation of WFS1 gene exon 8 in Iranian patients with Wolfram syndrome
International Journal of Diabetes in Developing Countries (2016)
-
Targeted genomic capture and massively parallel sequencing to identify novel variants causing Chinese hereditary hearing loss
Journal of Translational Medicine (2014)
