
Abstract
Cytokines are major immune system regulators. Previously, innate cytokine profiles determined by lipopolysaccharide stimulation were shown to be highly heritable. To identify regulating genes in innate immunity, we analyzed data from a genome-wide linkage scan using microsatellites in osteoarthritis (OA) patients (The GARP study) and their innate cytokine data on interleukin (IL)-1β, IL-1Ra, IL-10 and tumor necrosis factor (TNF)α. A confirmation cohort consisted of the Leiden 85-Plus study. In this study, a linkage analysis was followed by manual selection of candidate genes in linkage regions showing LOD scores over 2.5. An single-nucleotide polymorphism (SNP) gene tagging method was applied to select SNPs on the basis of the highest level of gene tagging and possible functional effects. QTDT was used to identify the SNPs associated with innate cytokine production. Initial association signals were modeled by a linear mixed model. Through these analyses, we identified 10 putative genes involved in the regulation of TNFα. SNP rs6679497 in gene CD53 showed significant association with TNFα levels (P=0.001). No association of this SNP was observed with OA. A novel gene involved in the innate immune response of TNFα is identified. Genetic variation in this gene may have a role in diseases and disorders in which TNFα is closely involved.
Similar content being viewed by others

Introduction
The immune system is a complex network of interacting pathways and signaling proteins that enables organisms to respond to pathogens and to many other events that challenge homeostasis. The immune system is regulated through cytokines, which are mainly secreted by lymphocytes. The ability of lymphocytes to produce cytokines can be characterized by ex vivo stimulation with, for example, the bacterial surface molecule lipopolysaccharide (LPS). This ligand triggers ex vivo lymphocytes to produce a maximal proinflammatory cytokine response, subsequently followed by an anti-inflammatory response.1 The as-such-determined maximal cytokine production profile has been shown to be stable2 and to contain a significant heritable component estimated between 53 and 86%, indicating a strong genetic control.3
On the basis of innate ex vivo cytokine profiles, individuals can be characterized as pro or anti-inflammatory and it has been shown that these profiles can predispose to diseases with an inflammatory component such as multiple sclerosis and cardiovascular events.4, 5 In osteoarthritis (OA), a proinflammatory profile may affect the articular cartilage homeostasis, which depends on a delicate balance between catabolic and anabolic activity induced, respectively, by pro- (tumor necrosis factor (TNF)α, interleukin (IL)-1β) and anti-inflammatory (IL-10 and IL-1 receptor antagonist (Ra)) cytokines.6, 7 In line with this hypothesis, Riyazi et al8 showed that patients of the Genetics osteoARthritis and Progression (GARP) study, with OA at multiple joint sites simultaneously, have high innate IL-1β and IL-1Ra and low innate IL-10 production compared with controls. Although a later study indicated that the mechanism underlying this association may be more complex, it confirmed the association of genetic variation of the innate cytokine levels with OA features.9 We, along with others, have shown that genetic variation of genes involved in the regulation of the immune system may be reflected by a specific profile of circulating plasma inflammatory markers.10, 11, 12 Furthermore, it was shown that DNA variants within the IL10 gene and genes of the IL-1 cluster may be responsible for a part of the variation in the heritable innate ex vivo cytokine production on LPS stimulation.13, 14, 15, 16 However, a large part of the heritability cannot be explained by the currently known genes.
Characterization of the genes that explain a considerable part of the individual variation in the innate cytokine profiles may shed more light on the regulatory elements designed to obtain or maintain a proper balance of these cytokines. Through a better understanding of these elements, more insight into the underlying disease processes in diseases with an inflammatory component such as OA can be obtained, thereby enabling the identification of putative therapeutic targets. In this study, we set off to discover such putative quantitative trait loci for innate cytokine levels using the available genome-wide linkage data of subjects of the GARP study,17 as well as data on their ex vivo LPS-stimulated production of IL-1β, IL-1Ra, IL-10 and TNFα.8 Innate cytokine levels were confirmed by association analysis using 563 unrelated individuals of the Leiden 85-Plus study.18 Identified genetic variation influencing the innate immunity profile was tested for association with OA and radiographic OA subtypes, as assessed in the GARP study.
Materials and methods
Study subjects
The GARP study consists of 191 Caucasian sibling pairs affected predominantly by symptomatic OA at multiple joint sites. Characteristics of the GARP study are listed in Table 1. Details of the phenotype and data collection are described by Riyazi et al.19 As a confirmation cohort, we used the Leiden 85-Plus study, which consists of inhabitants of Leiden (Netherlands), who were asked to participate in this study on reaching the age of 85 years between 1 September 1997 and 1 September 1999. The response rate was 87% and in total 599 individuals were included in this study. OA data are not available for the Leiden 85-Plus study.20
Phenotyping
For most participants (N=370 and 563, respectively) in the GARP study and Leiden 85-Plus study, an ex vivo whole-blood sample was stimulated with 10-ng/ml LPS, and, after a 4 h incubation, the sample was centrifuged and the TNFα levels were determined in the supernatant using an enzyme-linked immunosorbent assay. In a second sample, a similar protocol was performed with a 24 h incubation, after which the plasma levels of IL-1β, IL-1Ra and IL-10 were determined.1, 19 In concordance with previous studies,3, 4, 5 the ex vivo-LPS-stimulated cytokine levels were not normally distributed and influenced by gender. In our analyses, log-transformed cytokine levels were used and analyses were adjusted for sex.
Genotyping
Previously, a genome-wide microsatellite scan was performed in the GARP study to identify new OA susceptibility loci; a detailed description of the genotyping methods and control policy has been described by Meulenbelt et al.17 In short, 417 microsatellite markers on an average spacing of 10 cM across the genome were measured. Initial linkage peaks were identified and two peaks were fine mapped by typing three additional microsatellite markers for each in the region of linkage (Supplementary Table 1). Single-nucleotide polymorphism (SNP) multiplex genotyping assays were designed using Assay Designer software 3.1. iPlex assays were measured on the Sequenom MassARRAY system (Sequenom, San Diego, CA, USA). PCRs were carried out in a final volume of 5 μl and contained standard reagents and 5 ng of dried genomic DNA. Genotypes were called using the Genotyper v3.1 software (Sequenom). All SNPs were checked for deviations from Hardy–Weinberg equilibrium and approximately 8% of the subjects were genotyped twice as a check for genotyping and calling consistency. Of the 47 genotyped SNPs in this study, three SNPs failed quality check because of low amplification, bad cluster separation or low confidence in called genotypes, and were excluded from further analysis.
Linkage analysis and candidate gene selection
The GARP microsatellite genotype data and log-transformed LPS-stimulated levels were analyzed using the variance components option implemented in Merlin to assess linkage of the levels to genetic loci.21 Merlin output files were modified to tab delimited files with LOD score per marker to facilitate uploading to a custom track in the UCSC genome browser genome graph function. The significance level of linkage peaks was assessed using random gene dropping simulations in Merlin using 5000 reruns. The regions showing LOD scores over 2.5 were explored for candidate genes using the UCSC genome browser,22 where the individual markers’ LOD scores were uploaded on a custom track. All UCSC-annotated genes in the 1-LOD-drop region within the flanking areas of a linkage peak over 2.5 were considered for possible involvement in the cytokine response. This manual selection of genes was based on location within the linkage region, GO terms and Swissprot description provided in the UCSC genome graph function. Genes selected were those that are described as being involved in immune system communication, antigen recognition and immune response. We selected 10 positional candidate genes (three linkage areas). Candidate genes were subsequently tagged using SNPs selected from the International HapMap Project genome browser.23 SNP selection was based on genetic position and function as well as on the potential to tag genetic variation present within these genes. Tagger software implemented in the Haploview program with settings ‘r2>0.8’ and ‘pairwise tagging’ was used to optimize tagging SNP selection.24 In the selection process, we included only SNPs with minor allele frequencies over 0.05 in CEPH data. SNPs in coding regions were prioritized by forced inclusion of nonsynonymous SNPs and the lowest priority was given to downstream SNPs. In total, 47 SNPs were selected for genotyping.
Association analysis of quantitative innate cytokine levels
The GARP data were analyzed for association of LPS levels to SNPs in the candidate genes using QTDT.25 Initially, −WEGA and –WEGD commands were applied to test for association on the basis of linkage to a specific locus under an additive and dominant model, respectively. To test for possible population stratification, we used the –AP –WEGA or –AP −WEGD command. Furthermore, a linear mixed model was used to model the association with the SNP in the GARP and Leiden 85-Plus data including sex, age and BMI as covariates. Here, the random family effect shows correlation between siblings of the GARP study due to shared genetic and environmental effects.26 In the combined GARP and Leiden 85-Plus analysis, in addition to family numbers and covariates sex, age and BMI, we included study identifiers in the model to correct for putative batch differences. Genotypes were coded as 0 (homozygote common allele), 1 (heterozygote) and 2 (homozygote rare allele) to test an additive model and 0, 1 and 1 to test a dominant model with one degree of freedom. All reported P-values are nominal P-values uncorrected for multiple testing for the reader's interpretation of the results, unless mentioned otherwise.
Qualitative association analysis of OA status and SNPs
To assess the association of SNPs to OA at multiple joint sites as defined in the GARP study, a logistic regression was performed in STATA/SE 8.0, using the Leiden 85-Plus sample as a reference sample, whereby we used family numbers in the GARP study as a random effects variable to model familial dependencies. Dominant effects for the rare allele were tested by pooling heterozygotes with homozygotes for the rare allele.
RESULTS
Characteristics
The characteristics of GARP and the study sample of the Leiden 85-Plus study, in which both innate cytokine production levels and genotypes were available, are shown in Table 1. The participants of the Leiden 85-Plus study were significantly older compared with the GARP participants (P<0.01), and we observed significant differences in the transformed LPS-stimulated levels between the GARP and Leiden 85-Plus participants for IL-1β (P=0.034), IL-1Ra (P<0.01) and TNFα (P<0.01). To check for age dependencies of the LPS-stimulated cytokine profiles, the correlation with age was analyzed. We observed no significant correlations of age and IL-1β, Il-10 or IL-1Ra. However, TNFα showed a significant (P=0.037) correlation with age with a Pearson correlation coefficient of 0.11 (data not shown). The older subjects of the GARP study had on an average a higher LPS-stimulated TNFα level.
IL-1β, IL-1Ra and IL-10 linkage and association analysis
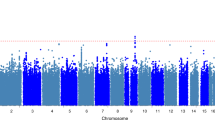
The genome-wide linkage analysis to find quantitative trait loci involved in innate IL-1β, IL-1Ra and IL-10 analyses using variance components did not reveal any evidence for linkage above an LOD score of 2.5 (Figure 1a–c, respectively). We did not select candidate genes and SNPs for follow-up analysis of these traits. No substantial evidence for linkage was observed at the loci encoding cytokines IL-1β and IL-1Ra (2q13), IL-10 (1q32.1) or TNFα (6p21.33) (Figure 1a–d).

LOD scores for genome-wide linkage analyses for QTLs of (a) IL-1β, (b) IL-1Ra, (c) IL-10 and (d) TNFα.
TNFα linkage and association analysis
Genome-wide linkage analysis of innate TNFα levels revealed three regions with a positive evidence for linkage with LOD scores over 2.5 (Figure 1d), of which one peak reached a genome-wide linkage significance level. The linkage peak on chromosome 11q12.1 (Figure 1d, peak 2) was fine mapped using three microsatellite markers, and after fine mapping showed a maximum LOD score of 2.57 (marker D11S1314, P=3.0 × 10−4, genome-wide P-value 0.497) between markers D11S935 and D11S901 (width 53 Mb, Figure 2a). Furthermore, on chromosome 17p13.2 (Figure 1d, peak 3), we observed a narrow linkage peak with a maximum LOD score of 3.38 (marker D17S938, P=4.0 × 10−5, genome-wide P-value 0.064) between markers D17S831 and D17S799 (width 5 Mb, Figure 2b), which was not fine mapped. The largest peak on chromosome 1p13.3 (Figure 1d, peak 1) was fine mapped using three additional microsatellite markers and after fine mapping showed a maximum LOD score of 3.77 (marker D1S2726, P=3.0 × 10−4, genome-wide P-value 0.018) between markers D1S2868 and D1S484 (width 52 Mb, Figure 2c). Within these linkage peaks, we selected 10 candidate genes (Table 2). On chromosome 11, we selected candidate genes MADD, SELH, CD6 and CD5 on chromosome 17 GPS2, TNFA-SF12/13 and CD68 and on chromosome 1 CSF1, CD53 and FAM19A3. The genes were tagged using 44 haplotype tagging SNPs, thereby tagging from 30 to 100% of the genotypic variation recorded in the HapMap database (Table 2). QTDT analysis indicated significant associations with TNFα levels for SNPs in CD53 and FAM19A3 (Table 2). We were unable to model the observed associations of FAM19A3 in a linear mixed model; however, when a dominant linear mixed model was fitted for CD53 rs6679497, we again observed a significant association in both the GARP and Leiden 85-Plus studies separately (P=0.013 and 0.032, respectively, Table 3). When we combined the data of both studies in a linear mixed model using a dominant model, we observed a highly significant association of rs6679497 (P=0.001, Table 3). The association remains significant after a Bonferroni correction to account for the 11 SNPs tested on the locus (corrected P-value=0.012), or accounting for all 44 SNPs selected on the linkage analysis (corrected P-value =0.047). The minor allele of this intron SNP (frequency 0.48) associated with significantly lower innate TNFα levels.

A detailed view of the initial and fine-mapped linkage peaks identified on chromosome 11 (panel a, peak 2), chromosome 17 (panel b, peak 3) and chromosome 1 (panel c, peak 1). Schematically represented are the tested gene positions in the linkage area. Dotted lines represent the initial linkage signal, whereas solid lines represent the fine-mapped linkage signal.
Association analysis of rs6679497 to OA
No significant association was observed for CD53 rs6679497 when GARP subjects (cases) were compared with subjects of the Leiden 85-Plus study as controls using the dominant model (adjusted for age, sex and BMI, P=0.142), indicating that the TNFα QTL locus did not confer susceptibility to OA.
Discussion and Conclusion
Through a genome-wide linkage scan, we identified SNP rs6679497 in CD53, of which the minor allele associates with lower innate TNFα levels. It can be hypothesized that the specific genotype of rs6679497 predisposes or protects its carriers from diseases and disorders in which TNFα has a substantial role. Previously, it was shown that TNFα does not have a major role in the onset of OA8 and in line with this hypothesis, we were unable to show associations of rs6679497 with OA as defined in the GARP study.
CD53 codes for cluster of differentiation 53, a leukocyte surface antigen. The protein family to which this cell surface glycoprotein belongs to is known to complex with integrins, cellular components involved in cell–cell and cell–matrix interactions. CD53 deficiency has been linked to recurring infectious diseases caused by bacteria, fungi and viruses;27 susceptibility to these might be increased for carriers of the minor allele of rs6679497. The protein is implicated in elevated cellular glutathione in response to LPS activation and may increase cell survival under UV-B and oxidative conditions.28 Furthermore, treatment of neutrophils with TNFα downregulates the presence of CD53 antigens on the cell surface through a proteolytic mechanism.29 This indicates that the protein may have a substantial role in cellular stability and in the inflammatory response to adverse conditions. Furthermore, the protective effect of ligated CD53 on the cellular surface may help specific tumors to escape from programed cell death.30 Although rs6679497 or any of the SNPs in its LD block are investigated for effects on the expression levels or protein function, the gene is under strong genetic control,31 and genetic variation might have a role in tumor biology or other diseases. Such a relation can readily be elucidated through investigation of this marker SNP in cancer cohorts. Furthermore, in a study that characterizes leukocytes from normal and rheumatoid arthritis (RA) patients, CD53 was found to be elevated on the RA lymphocytes surfaces.32 Investigating the role of this gene in relation to this and other TNFα-driven diseases later in life may show protective effects of the rs6679497 minor allele.
The SNP CD53 rs6679497 resides within the intron of the gene, which shows low levels of conservation across species, and it is in an LD block across several introns and exons encompassing at least 23 other intron SNPs (recorded in the HapMap phase 1 and 2 data). Several transcripts are known as recorded in the UCSC database (accession number ENSG00000143119); however, only a proportion of these will actually be translated into a protein. Given the current level of total variation tagging (52%), it is likely that the SNP is only a proxy marker in LD with a causal (functional) polymorphism, which could have a more obvious implication on gene regulation or protein stability and functioning. This is substantiated by the fact that the CD53 SNP explains only a part of the linkage as determined in QTDT analyses (results not shown). To find the true functional variant, a more detailed analysis of this gene by sequencing or SNP saturation is necessary. Alternatively, in our candidate gene approach, we may have missed additional genetic variation at the loci of interest because of a knowledge bias on both the presence and role of genes at these loci.
Although it was shown previously that IL1 cluster haplotypes were associated with IL-1β bioavailability,33, 34 we observed no evidence of linkage on the genetic loci for the respective cytokines’ genes, the IL-1 gene cluster on chromosome 2, IL-10 on chromosome 1 or TNF on chromosome 6. Possibly, the genetic variation in LPS-stimulated cytokine levels explained by these loci is not readily detected by linkage analysis, which is known to be most suitable to detect loci that explain a major part of genetic variation. Furthermore, our linkage analyses of innate IL-1β, IL-1Ra and IL-10 levels in general revealed only moderate linkage peaks up to an LOD score of 2.5. More likely, as was shown in a previous study,33 the estimate of innate IL-1β production on LPS stimulation may not be entirely independent of the OA disease status, possibly by sensitization of the Toll-like receptor pathways as a result of disease activity35 or otherwise sensitization of the response by lymphocytes, which may have interfered with the current linkage analysis by introducing cohort heterogeneity or bias. In the linkage analyses, we checked whether the levels in the GARP study sample were normally distributed to facilitate powerful linkage analysis using the variance component option. The innate levels of IL-1β, IL-10 and TNFα were normally distributed, whereas innate IL-1Ra levels were normally distributed after removal of one extreme value, which did not alter the linkage analysis results (data not shown). The use of Merlin–Regress,21 which may be more appropriate for use in highly selected samples, showed a similar pattern of LOD scores for all traits, with slightly lower maximum LOD scores. It remains possible that the observed association only occurs in the middle aged and elderly; therefore, further searches for genetic loci that influence ex vivo innate cytokine profiles may benefit from the use of healthy young subjects in these searches.
Following a genome-wide linkage analysis, namely, an association analysis of positional candidate gene SNPs within the 1-LOD-drop interval of a linkage peak, we show a consistent association of SNP rs6679497 in CD53 with innate TNFα levels in both the GARP study (P=0.013) and the confirmation cohort consisting of Leiden 85-Plus participants (P=0.032). A dominant linear mixed model analysis on combined data from the GARP and Leiden 85-Plus study showed that the minor allele of this SNP associated with a highly significantly lower innate TNFα level independent of age and sex. In diseases with a large TNFα component, such as inflammatory bowel disease or rheumatoid disorders, the minor allele of rs6679497 might exert a protective effect in susceptibility or severity.
Accession codes
References
Ohlsson K, Linder C, Lundberg E, Axelsson L : Release of cytokines, proteases from human peripheral blood mononuclear, polymorphonuclear cells following phagocytosis, LPS stimulation. Scand J Clin Lab Invest 1996; 56: 461–470.
van der Linden MW, Huizinga TW, Stoeken DJ, Sturk A, Westendorp RG : Determination of tumour necrosis factor-alpha, interleukin-10 production in a whole blood stimulation system: assessment of laboratory error, individual variation. J Immunol Methods 1998; 218: 63–71.
de Craen AJ, Posthuma D, Remarque EJ, van den Biggelaar AH, Westendorp RG, Boomsma DI : Heritability estimates of innate immunity: an extended twin study. Genes Immun 2005; 6: 167–170.
de Jong BA, Schrijver HM, Huizinga TW et al: Innate production of interleukin-10, tumor necrosis factor affects the risk of multiple sclerosis. Ann Neurol 2000; 48: 641–646.
McGeer PL, McGeer EG : Innate immunity, local inflammation, degenerative disease. Sci Aging Knowledge Environ 2002; 2002: re3.
Goldring MB : Update on the biology of the chondrocyte, new approaches to treating cartilage diseases. Best Pract Res Clin Rheumatol 2006; 20: 1003–1025.
Goldring MB, Otero M, Tsuchimochi K, Ijiri K, Li Y : Defining the roles of inflammatory, anabolic cytokines in cartilage metabolism. Ann Rheum Dis 2008; 67 (Suppl 3): iii75–iii82.
Riyazi N, Slagboom E, de Craen AJ et al: Association of the risk of osteoarthritis with high innate production of interleukin-1beta, low innate production of interleukin-10 ex vivo, upon lipopolysaccharide stimulation. Arthritis Rheum 2005; 52: 1443–1450.
Chapman K, Loughlin J : Association of the interleukin-1 gene cluster with osteoarthritis of the hip: comment on the article by Meulenbelt et al, the letter by Smith et al. Arthritis Rheum 2006; 54: 3722–3723.
Bos SD, Suchiman HE, Kloppenburg M et al: Allelic variation at the C-reactive protein gene associates to both hand osteoarthritis severity, serum high sensitive CRP levels in the GARP study. Ann Rheum Dis 2007; 67: 877–879.
Curran JE, Jowett JB, Elliott KS et al: Genetic variation in selenoprotein S influences inflammatory response. Nat Genet 2005; 37: 1234–1241.
Rafiq S, Frayling TM, Murray A et al: A common variant of the interleukin 6 receptor (IL-6r) gene increases IL-6r and IL-6 levels, without other inflammatory effects. Genes Immun 2007; 8: 552–559.
Haukim N, Bidwell JL, Smith AJ et al: Cytokine gene polymorphism in human disease: on-line databases, supplement 2. Genes Immun 2002; 3: 313–330.
Danis VA, Millington M, Hyland VJ, Grennan D : Cytokine production by normal human monocytes: inter-subject variation and relationship to an IL-1 receptor antagonist (IL-1Ra) gene polymorphism. Clin Exp Immunol 1995; 99: 303–310.
Wen AQ, Wang J, Feng K, Zhu PF, Wang ZG, Jiang JX : Effects of haplotypes in the interleukin 1beta promoter on lipopolysaccharide-induced interleukin 1beta expression. Shock 2006; 26: 25–30.
Temple SE, Lim E, Cheong KY et al: Alleles carried at positions -819 and -592 of the IL10 promoter affect transcription following stimulation of peripheral blood cells with Streptococcus pneumoniae. Immunogenetics 2003; 55: 629–632.
Meulenbelt I, Min JL, Bos S et al: Identification of DIO2 as new susceptibility locus for symptomatic Osteoarthritis. Hum Mol Genet 2008; 17: 1867–1875.
van Exel E, Gussekloo J, Houx P et al: Atherosclerosis and cognitive impairment are linked in the elderly. The Leiden 85-plus Study. Atherosclerosis 2002; 165: 353–359.
Riyazi N, Meulenbelt I, Kroon HM et al: Evidence for familial aggregation of hand, hip, and spine but not knee osteoarthritis in siblings with multiple joint involvement: the GARP study. Ann Rheum Dis 2005; 64: 438–443.
van Exel E, Gussekloo J, de Craen AJ, Bootsma-van der WA, Frolich M, Westendorp RG : Inflammation and stroke: the Leiden 85-Plus Study. Stroke 2002; 33: 1135–1138.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR : Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101.
Kuhn RM, Karolchik D, Zweig AS et al: The UCSC Genome Browser Database: update 2009. Nucleic Acids Res 2009; 37: D755–D761.
Frazer KA, Ballinger DG, Cox DR et al: A second generation human haplotype map of over 3.1 million SNPs. Nature 2007; 449: 851–861.
Goode EL, Fridley BL, Sun Z et al: Comparison of tagging single-nucleotide polymorphism methods in association analyses. BMC Proc 2007; 1 (Suppl 1): S6.
Abecasis GR, Cardon LR, Cookson WO : A general test of association for quantitative traits in nuclear families. Am J Hum Genet 2000; 66: 279–292.
Diggle PJ, Liang KY, Zeger SL : General linear models for longitudinal data. In: Atkinson AC, Copas JB, Pierce DA, Schervish MJ, Titterington DM (ed). Analysis of Longitudinal Data. Oxford University Press: Oxford, 1994; pp 68–77.
Mollinedo F, Fontan G, Barasoain I, Lazo PA : Recurrent infectious diseases in human CD53 deficiency. Clin Diagn Lab Immunol 1997; 4: 229–231.
Kim TR, Yoon JH, Kim YC et al: LPS-induced CD53 expression: a protection mechanism against oxidative and radiation stress. Mol Cells 2004; 17: 125–131.
Mollinedo F, Martin-Martin B, Gajate C, Lazo PA : Physiological activation of human neutrophils down-regulates CD53 cell surface antigen. J Leukoc Biol 1998; 63: 699–706.
Yunta M, Lazo PA : Apoptosis protection and survival signal by the CD53 tetraspanin antigen. Oncogene 2003; 22: 1219–1224.
Hernandez-Torres J, Yunta M, Lazo PA : Differential cooperation between regulatory sequences required for human CD53 gene expression. J Biol Chem 2001; 276: 35405–35413.
Pedersen-Lane JH, Zurier RB, Lawrence DA : Analysis of the thiol status of peripheral blood leukocytes in rheumatoid arthritis patients. J Leukoc Biol 2007; 81: 934–941.
Smith AJ, Keen LJ, Billingham MJ et al: Extended haplotypes and linkage disequilibrium in the IL1R1-IL1A-IL1B-IL1RN gene cluster: association with knee osteoarthritis. Genes Immun 2004.
Moxley G, Han J, Stern AG, Riley BP : Potential influence of IL1B haplotype and IL1A-IL1B-IL1RN extended haplotype on hand osteoarthritis risk. Osteoarthritis Cartilage 2007; 15: 1106–1112.
Scanzello CR, Plaas A, Crow MK : Innate immune system activation in osteoarthritis: is osteoarthritis a chronic wound? Curr Opin Rheumatol 2008; 20: 565–572.
Acknowledgements
We acknowledge the support of the following cooperating hospitals and referring rheumatologists, as well as the orthopedic surgeons and general practitioners in our region, for identifying eligible GARP patients, in random order: Dr LNJEM Coene, Department of orthopedic surgery and Dr HK Ronday, Department of rheumatology, Leyenburg Hospital, the Hague; I Speyer and Dr ML Westedt, Department of rheumatology, Bronovo Hospital, the Hague; Dr D van Schaardenburg, Department of Rheumatology, Jan van Breemen, Institute in Amsterdam; Dr AJ Peeters and Dr D van Zeben, Department of rheumatology, Reinier de Graaf Hospital, Delft; Dr EJ Langelaan, Department of orthopedic surgery, Rijnland Hospital in Leiderdorp; and Dr Y Groeneveld, general practitioner, associated with the Leiden University Medical Center. In addition to the grant support from The Dutch League against Rheumatism (NR 04-1-403), Pfizer Inc., Groton, CT, USA and the Center of Medical Systems Biology (CMSB) provided generous financial support for this work. The Leiden 85-plus Study is a collaborative project of the Department of Gerontology and Geriatrics (Rudi Westendorp, principal investigator) and the Department of Public Health and Primary Care (Jacobijn Gussekloo, principal investigator) of the Leiden University Medical Center, Leiden, the Netherlands.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Bos, S., Lakenberg, N., van der Breggen, R. et al. A genome-wide linkage scan reveals CD53 as an important regulator of innate TNF-α levels. Eur J Hum Genet 18, 953–959 (2010). https://doi.org/10.1038/ejhg.2010.52
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.52
Keywords
This article is cited by
-
Identification of key genes as predictive biomarkers for osteosarcoma metastasis using translational bioinformatics
Cancer Cell International (2021)
-
New immunological potential markers for triple negative breast cancer: IL18R1, CD53, TRIM, Jaw1, LTB, PTPRCAP
Discover Oncology (2021)
-
Bioinformatics analysis reveals novel hub gene pathways associated with IgA nephropathy
European Journal of Medical Research (2020)
-
Association between CD53 genetic polymorphisms and tuberculosis cases
Genes & Genomics (2019)
-
Pathogen lineage-based genome-wide association study identified CD53 as susceptible locus in tuberculosis
Journal of Human Genetics (2017)
