
Abstract
Essential hypertension is a major cardiovascular risk factor and a large proportion of this risk is genetic. Identification of genomic regions consistently associated with hypertension has been difficult in association studies to date as this requires large sample sizes.
We previously published a large genome-wide linkage scan in Americans of African (AA) and European (EA) descent in the GenNet Network of the Family Blood Pressure Program (FBPP). A highly significant linkage peak was identified on chr1q spanning a region of 100 cM. In this study, we genotyped 1569 SNPs under this linkage peak in 2379 individuals to identify whether common genetic variants were associated with blood pressure (BP) at this locus.
Our analysis, using two different family-based association tests, provides suggestive evidence (P≤2 × 10−5) for a collection of single nucleotide polymorphisms (SNPs) associated with BP. In EAs, using diastolic BP as a quantitative phenotype, three variants located in or near the GPA33, CD247, and F5 genes, emerge as our top hits; for systolic BP, variants in GPA33, CD247, and REN are our best findings. No variant in AAs came close to suggestive evidence after multiple-test corrections (P≥8 × 10−5).
In summary, we show that systematic follow-up of a linkage signal can help discover candidate variants for essential hypertension that require a follow-up in yet larger samples. The failure to identify common variants is either because of low statistical power or the existence of rare coding variants in specific families or both, which require additional studies to clarify.
Similar content being viewed by others

Background
Essential hypertension is quantitatively the major risk factor for premature cardiovascular disease and stroke.1 Worldwide, from 5.6% (low- and middle-income countries) to 9.3% (high-income countries) of the total disease burden is estimated to be a consequence of high blood pressure (BP).2 Although hypertensive patients can be treated to lower BP, and this treatment reduces the rate of cardiovascular complications,3 the pathogenesis of essential hypertension remains unclear.
Given the ∼45% heritability of essential hypertension, genetic analysis is one contemporary approach to learn about the molecular origins of the disease. This assumption has been the driving force for large consortia to investigate the genetic basis of essential hypertension, such as the National Heart, Lung, and Blood Institute funded Family Blood Pressure Program (FBPP).4 The GenNet network within the FBPP has previously published results of a genome-wide linkage study on African-American (AA) and European-American (EA) participants. The most compelling evidence for linkage was found on chromosome 1q23–q32.5 We have subsequently extended the linkage analysis (LOD score ∼4.3) by adding additional microsatellite markers in this region and also genotyped 58 SNPs in candidate genes under the linkage peaks in 816 AA and 1010 EA participants.6 We now present results of a comprehensive family-based association study using genotypes from 1569 SNPs in this region, performed with a customized molecular inversion probe microarray.7, 8, 9 The genotyped variants cover the entire linkage peak and were genotyped in an extended sample of 2379 individuals.
Methods
Patient recruitment and phenotype ascertainment
GenNet recruited African-American (n=1101) and European-American participants (n=1497) at two field-centers between 1995 and 2003, based on a hypertensive proband. Non-Hispanic white subjects were recruited from Tecumseh, Michigan, and African-American subjects were recruited from Maywood, Illinois. Probands were defined as individuals aged 18–50 years with blood pressures in the upper 20th to 25th percentile of the age/gender-specific blood pressure distribution. Once the proband was identified, an attempt was made to enroll all siblings and parents of the proband, irrespective of their blood pressure or hypertension treatment status. The hypertensive proband was included in the analysis. The respective institutional review boards approved the study and all individuals included gave written informed consent.4
BP measurements were carried out according to standard procedures in a sitting position after a resting period.4 Subjects were not allowed to smoke or drink coffee before the visit. For each reading, the pressure at Korotkoff phase I sound was taken as the SBP. DBP was determined at the Korotkoff phase IV sound in children aged less than 13 years and at the Korotkoff phase V sound in all other subjects. The average of two manual BP measurements was used as a phenotype. Of all 2598 participants, DNA was available on 2413 individuals (92.9%). Individuals treated by antihypertensive drugs had their blood pressure values corrected by adding 10 and 5 mm Hg to their SBP and DBP, respectively where indicated.
Study design
Fine-mapping linkage analysis yielded one major and two minor linkage peaks in a total region of 100 cM.6 We selected three genomic regions directly under the major and two minor peaks. All coordinates and gene names mentioned are on chromosome 1 and refer to NCBI Build 36.1, for details of coordinates see Online Supplementary Information. Within these three regions, totaling 9 031 708 bp, 1800 SNPs were chosen with the following criteria: we started with all SNPs in HapMap phase II (release 19) with minor allele frequency between 0.1 and 0.9 for Yorubans (YRI) and Whites of European Origin (CEU). We eliminated all SNPs present, and in complete LD (r 2=1) with the Affymetrix Mapping 500 K array. We chose to exclude the Affymetrix 500 K SNPs because we anticipated that these would become available in the contemplated genome-wide association studies. Tagging SNPs were selected using the following selection thresholds: r 2≥0.8, LOD 3.0 for 2-marker haplotypes. Using these selection criteria, we chose 1332, 10, and 322 SNPs for regions 1, 2, and 3, respectively. A small number of other SNPs were added (see Online Supplementary Information).
Bioinformatics analysis by Affymetrix converted 1721 of the 1800 SNPs into reliable assays on an Affymetrix GeneChip Targeted Genotyping 3 K microarray (MIP array in the following). A full list with all 1721 SNPs is provided in the Online Supplementary Information (Supplementary Table S1).
Genotyping
The MIP arrays were run in batches of 24 or 48 samples according to the manufacturer's instructions, with the exception that we used half the DNA and reagent amounts in the initial reaction. This had no impact on quality metrics. For details see Online Supplementary Information.
Genotype calling
Genotype calling was performed on a Dell desktop workstation, using the Affymetrix GeneChip TG software (Affymetrix, SantaClara, CA, USA.9 All microarrays having passed the manufacturer's quality control genotype calling were subjected to one single clustering and genotype calling. To test the quality metrics of the genotypes obtained, we ran the clustering and genotyping procedures over a range of each of the two main quality cutoff values: minimum call confidence (range: 0–100%) and the minimum call rate (range: 0–100%). The resulting curve of reproducibility vs completeness (Supplementary Figure S1) shows that even at maximal data completeness (98.8%), accuracy estimated by reproducibility is very high (99.5%). Given these metrics, we used the most complete dataset for the genetic analysis. The SNP inclusion criteria applied to this dataset were: per SNP call rate of ≥0.95, per SNP Hardy–Weinberg equilibrium P≥10−4. Of the 1717 chromosome 1 SNPs typed, 148 were removed for not satisfying per SNP call rate and Hardy–Weinberg equilibrium criteria in one or both of the ethnicities.
Quality control and analysis methods used
Three of the genes in the target region, RGS5, ATP1B1, and SELE, have been previously analyzed by genotyping 58 SNPs by Taqman and several variants were associated with SBP and DBP.6 We repeated genotyping for all 58 SNPs in this experiment: the MIP and Taqman genotypes showed a concordance of 0.996. Compared to the previous experiment with 58 SNPs, the mean per SNP call rate is higher in this comprehensive survey (99.7 vs 97.1%), the sample size is larger for both ethnicities (938 vs 816 for AA; 1441 vs 1010 for EA), and some pedigree errors (mainly joining of families into more complete family structures) have since been corrected. For these reasons, identical association results for the 58 SNPs were not obtained, but not expected either (see Online Supplementary Information, Supplementary Table S2 for details).
Statistical methods for genetic analysis
Data file preparation, data quality control, descriptive statistics, and data display were carried out using the Haploview implementation of Tagger,10 the PLINK analysis toolset,11 custom code written in R version 2.7.0 (The R Foundation for Statistical Computing), and custom Perl scripts. Where indicated, the following regression of the phenotype on age and sex was used and the residuals utilized for further analysis: y=α+β1 × age(years)+β2 × sex(1 for male, 2 for female)+ɛ.
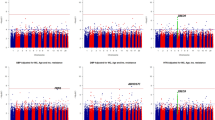
To maximize statistical power, we used quantitative phenotypes in the following analysis. We stratified the analysis by ethnicity (AA and EA); systolic (SBP) and diastolic (DBP) blood pressures were analyzed separately. For each of SBP and DBP, the association was tested for three closely related variables: (1) raw measured mean SBP/DBP without correction for antihypertensive treatment, (2) raw measured SBP/DBP only for individuals without antihypertensive medication (excluding all values from individuals on BP-lowering drugs), and (3) the SBP/DBP to which a correction factor for antihypertensive treatment had been applied. The two major confounders for BP are age and sex. We analyzed both age/sex-corrected phenotypes and uncorrected phenotypes separately. We did not use BMI adjustment because our previous linkage analysis was done without regression on BMI. We used two different tools for family-based association studies: For regression-based tests of association, the MERLIN software package (– assoc option)12 was used; for TDT-based tests, the FBAT software package (hbat –p option) (Abecasis Laboratory, University of Michigan, Ann Arbor, USA)13 was used. The family-based association test from the MERLIN software package12 assumes an additive model. FBAT was set to an additive model. The ‘10/5 mm Hg method’ was used as a correction method to adjust for the presence of blood pressure-lowering treatment where indicated, a method shown to perform well in simulation studies.14 Recombination rates (Figures 1 and 2, Supplementary Figures S2 and S3) for the genomic segment was obtained from HapMap phase II.15

P-value Manhattan plot for age and sex adjusted phenotypes in European-Americans: SBP phenotypes (a) and DBP (b). Nominal P-values for association with SBP and DBP are plotted by genomic position on chromosome 1 for each SNP tested. Phenotypes adjusted for age and sex were used for this analysis. The three regions assayed are depicted with different background colors (region 1: light green; region 2: light blue; region 3: light violet). The top panel shows P-values obtained with two family-based association tests (MERLIN in orange and FBAT in blue) and the bottom panel depicts the local recombination rate (cM/Mb).

P-value Manhattan plot for phenotypes without adjustment in European-Americans, same as in Figure 1, except that phenotypes used for this analysis were not adjusted for age and sex: SBP phenotypes (a) and DBP phenotypes (b).
We intended to use a conservative correction for multiple testing. The experiment-wide significance level was estimated for EA based on a Bonferroni-corrected α of P=0.05. We took into account: (1) the number of SNPs tested (excluding SNPs in high LD with each other), and (2) the effective number of phenotypes tested. In total 1569 SNPs were tested; of these, HapMap genotype data was available for 1541. Among these 1541 SNPs, 475 showed pairwise correlation with r2=0.6 or higher in EA, based on HapMap genotypes. We subtracted the 475 correlated SNPs from the total number of SNPs, resulting in 1094 effective tests for SNPs. All phenotypes tested are strongly correlated to each other. Therefore, although, two times three phenotypes were tested (three for SBP phenotypes and three for DBP phenotypes), they do not constitute six independent tests. The r2 between the variables are as follows: ‘SBP’ – ‘SBPuntreated’: 0.8 (80% of observations are identical, 20% are missing for ‘SBP untreated’); ‘SBP’ –‘SBP treatment corrected’: 0.96 (age and sex corrected) and 0.97 (uncorrected); ‘SBP’ – ‘DBP’: 0.37; ‘SBP’ – ‘age and sex corrected SBP’: 0.72; ‘DBP’ – ‘age and sex corrected DBP’: 0.88. We calculated the number of independent phenotypic tests according to the following formula: 1+(1−r2SBP−SBPuntreat)+(1−r2SBP−SBPtreatcorr)+[(1−r2SBP−DBP) × [1+(1−r2DBP−DBPuntreat)+(1−r2DBP−DBPtreatcorr)]]=2.02. Bonferroni correction of a type I error of 0.05 for 1094 SNPs and 2.2 phenotypic tests yields a significance threshold of 2.1 × 10−5. As we are using two testing methods and apply them to two ethnicities, this threshold reduces to 5.2 × 10−6. Given our experimental setup, it is difficult to determine a more exact significance level without many more assumptions and the precise experiment-wide significance level might thus be lower. The same significance threshold was applied to the AA samples, but has to be considered even more liberal here given the globally lower LD in AA.
Results
Study participant characteristics
Participants from both ethnicities were recruited in families (trios, duos, sibships, multi-generational families) based on a proband with high BP. All available 2413 samples were genotyped in this study and data was obtained on 2379 samples (938 AA and 1441 EA). Table 1 lists key epidemiologic characteristics of this sample. AA and EA belong to 364 and 434 families respectively, with a median age of 38 and 43 years (AA and EA, respectively) because the GenNet population is fairly young. The distribution of SBP and DBP is provided in the Online Supplementary Information (Supplementary Figure S4). Forty-nine and 51 percent of individuals are female and close to 20% of individuals are treated by BP-lowering medications.
Target region and genotyping QA & QC
We used a molecular inversion probe (MIP) assay7 to specifically target 1717 SNPs under the linkage peak consisting of one major peak and two minor peaks.6 The following analysis is based on 1569 SNPs that survived quality analysis. The total genomic region covered was 9 Mb and within this region coverage was at a median density of one SNP per 3.2 kb. The region covered contains 80 Refseq genes.
Association patterns in the target region on chromosome 1
Two analysis tools were used: MERLIN to maximize power as all samples can be used but this method is susceptible to population structure effects. FBAT is based on the transmission disequilibrium test (TDT) and avoids population structure effects but has lower power than MERLIN. Significance levels for every SNP and each phenotype using the two analysis methods are depicted in Figures 1 and 2 for EA. Figure 1 shows the P-values for phenotypes corrected for age and sex (same correction as for our previous linkage scan), and Figure 2 for uncorrected phenotypes. QQ plots for each analysis and the corresponding genomic control λ values are available in the Online Supplementary Information (Supplementary Figure S5 and Supplementary Table S3). For age- and sex-adjusted phenotypes a variant localized in intron 14 of the coagulation factor V precursor gene (F5) was most significant. In our analysis, using uncorrected phenotypes, the three following variants were most significant: a variant 5′ of the renin precursor gene (REN), a variant in intron 1 of the transmembrane glycoprotein A33 precursor gene (GPA33), and a variant in intron 1 of the T-cell receptor zeta chain precursor gene (CD247). Two of these genes also contained a variant that emerged among the top findings of the DBP phenotypes, for GPA33 it is the same SNP whereas for CD247 it is another SNP 28 kb apart. The genes identified, their variants, and the salient features of the analysis are summarized in Table 2.
The same analysis was conduced for the 938 AA participants genotyped for the same markers. No variant came close to the suggestive evidence as for EA; the smallest significance level was P=8 × 10−5 (cf. Supplementary Figures S2 and S3).
Discussion
We have conducted a detailed analysis of the main linkage peak for BP in GenNet, a network of the Family Blood Pressure Program (FBPP). Genotypes were obtained for 1569 SNPs in a 9-Mb region on chromosome 1 in 2379 Americans with African and European ancestry. For the 1441 EA participants, variants in four genes emerged as top hits for BP traits using a threshold of suggestive evidence based on the effective number of tests, the estimated number of phenotypes tested, the two analysis methods applied and the two ethnicities considered. For AA participants (n=938), no individual variant reached the threshold of suggestive evidence as defined in the EA.
BP is characterized by two primary measures, one during systole and the other during diastole. SBP is the peak pressure reached in the vascular system following cardiac contraction, DBP refers to the lowest pressure following the filling phase. Although the correlation of BP with cardiovascular risk is better for DBP in individuals younger than 50 years, SBP appears to be a better predictor for older individuals,16 and SBP and DBP are of course reasonably well correlated (r2=0.37 in GenNet). These similarities and distinctions are important because we analyzed both SBP and DBP phenotypes here, whereas the initial linkage peak, leading to the design of this study, was discovered using DBP in EA.5 Fine mapping of the linkage peak was carried out using age- and sex-corrected DBP as a phenotype and the strongest signal was detected including only participants of the study who did not receive BP-lowering therapy. It is striking that in the current study all top loci identified have at least one hit in the analysis of the untreated participants only.
Of the four genes associated with SBP and DBP in this study, two have previously been associated with essential hypertension (REN and F5). All variants identified are located in introns (see Table 2) or, for REN, 12.5 kb upstream of the renin precursor gene promoter.
Cleavage of the renin precursor (REN) yields the mature protein, which is a key regulator of BP through the renin—angiotensin–aldosterone system. It is also the target of a new medication for a BP-lowering therapy.17 Some variants have been described to be associated with BP and BP treatment response,18 but these findings could not be confirmed in other studies. To our knowledge rs11240692 has not been implicated in BP regulation or renin expression so far. Coagulation factor V precursor (F5) is a key player in the coagulation cascade, and rare and common mutations have been described leading to thrombophilia.19 It has been suggested that variant rs6025, the variant-defining factor V Leiden, is more frequent among individuals with essential hypertension.20 Transmembrane glycoprotein A33 precursor (GPA33) is a cell-surface antigen that is expressed on a great majority of human cancer cells.21 The mature protein product of the T-cell receptor zeta chain precursor (CD247) gene is an important immunologic cell-surface molecule, coupling antigen binding to intracellular messaging pathways.22 To our knowledge the latter two genes have not been associated with essential hypertension previously.
We had previously reported significant findings in the RGS5, ATP1B1, and SELE genes using genotype data from 58 SNPs genotyped by Taqman.6 The variants previously identified as significant are not significant in this study. The difference in the older and newer P-value is most likely due to multiple differences of a larger sample size, a higher call rate, and corrections of pedigree errors.
It is striking that our results in the large cohort of AA participants are overall far less significant (see Online Supplementary Information, Supplementary Figures S2 and S3). This has been observed in other studies as well23 and might be explained by the higher recombination rates in individuals with African ancestry or incomplete accounting of admixture across individuals. For future genetic investigations in individuals with African ancestry, this is clearly a question that requires more careful investigation.
In summary, this study shows that consistent follow-up of a major linkage peak on chromosome 1 yields several suggestive variants for SBP and DBP but no replication of the previous suggested variants or in other genes at this locus. This suggests that the statistical power for detecting the association is low despite a marker density comparable to current genome-wide genotyping arrays (median of 3 kb per SNP), probably due to small genetic effect sizes in this interval. It is also possible that the original linkage study results are explainable by rare variants at one or more loci in this interval that only deep and comprehensive sequencing can reveal.
References
Wilson PW : Established risk factors and coronary artery disease: the Framingham Study. Am J Hypertens 1994; 7: 7S–12S.
Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ : Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet 2006; 367: 1747–1757.
Staessen JA, Fagard R, Thijs L et al: Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. The Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet 1997; 350: 757–764.
Multi-center genetic study of hypertension: The Family Blood Pressure Program (FBPP). Hypertension 2002; 39: 3–9.
Thiel BA, Chakravarti A, Cooper RS et al: A genome-wide linkage analysis investigating the determinants of blood pressure in whites and African Americans. Am J Hypertens 2003; 16: 151–153.
Chang YP, Liu X, Kim JD et al: Multiple genes for essential-hypertension susceptibility on chromosome 1q. Am J Hum Genet 2007; 80: 253–264.
Hardenbol P, Baner J, Jain M et al: Multiplexed genotyping with sequence-tagged molecular inversion probes. Nat Biotechnol 2003; 21: 673–678.
Hardenbol P, Yu F, Belmont J et al: Highly multiplexed molecular inversion probe genotyping: over 10,000 targeted SNPs genotyped in a single tube assay. Genome Res 2005; 15: 269–275.
Moorhead M, Hardenbol P, Siddiqui F et al: Optimal genotype determination in highly multiplexed SNP data. Eur J Hum Genet 2006; 14: 207–215.
Barrett JC, Fry B, Maller J, Daly MJ : Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263–265.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR : Merlin – rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101.
Lange C, Silverman EK, Xu X, Weiss ST, Laird NM : A multivariate family-based association test using generalized estimating equations: FBAT-GEE. Biostatistics 2003; 4: 195–206.
Tobin MD, Sheehan NA, Scurrah KJ, Burton PR : Adjusting for treatment effects in studies of quantitative traits: antihypertensive therapy and systolic blood pressure. Stat Med 2005; 24: 2911–2935.
Frazer KA, Ballinger DG, Cox DR et al: A second generation human haplotype map of over 3.1 million SNPs. Nature 2007; 449: 851–861.
Franklin SS, Larson MG, Khan SA et al: Does the relation of blood pressure to coronary heart disease risk change with aging? The Framingham Heart Study. Circulation 2001; 103: 1245–1249.
Jensen C, Herold P, Brunner HR : Aliskiren: the first renin inhibitor for clinical treatment. Nat Rev Drug Discov 2008; 7: 399–410.
Moore N, Dicker P, O’Brien JK et al: Renin gene polymorphisms and haplotypes, blood pressure, and responses to renin-angiotensin system inhibition. Hypertension 2007; 50: 340–347.
Martinelli I, Bucciarelli P, Margaglione M, De Stefano V, Castaman G, Mannucci PM : The risk of venous thromboembolism in family members with mutations in the genes of factor V or prothrombin or both. Br J Haematol 2000; 111: 1223–1229.
Makris TK, Krespi PG, Hatzizacharias AN et al: Resistance to activated protein C and FV leiden mutation in patients with a history of acute myocardial infarction or primary hypertension. Am J Hypertens 2000; 13: 61–65.
Heath JK, White SJ, Johnstone CN et al: The human A33 antigen is a transmembrane glycoprotein and a novel member of the immunoglobulin superfamily. Proc Natl Acad Sci USA 1997; 94: 469–474.
Weissman AM, Baniyash M, Hou D, Samelson LE, Burgess WH, Klausner RD : Molecular cloning of the zeta chain of the T cell antigen receptor. Science 1988; 239: 1018–1021.
Ehret GB, Morrison AC, O’Connor AA et al: Replication of the Wellcome Trust genome-wide association study of essential hypertension: The Family Blood Pressure Program. Eur J Hum Genet 2008; 16: 1507–1511.
Acknowledgements
We thank all the volunteers who have participated in the FBPP studies (http://www.biostat.wustl.edu/fbpp/FBPP.shtml) and the NHLBI for funding. We thank Dr Maria Carolina Delgado at the University of Michigan and Dr Christy Chang, at the University of Maryland, for help with this study, and Dr Jeff Smith and Dr Meg Mintz from Affymetrix for outstanding support. We are indebted to the anonymous reviewers of this work that led to improvement of the presentation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Conflict of interest
Aravinda Chakravarti is a paid member of the Scientific Advisory Board of Affymetrix Inc., a potential conflict of interest managed by Johns Hopkins University School of Medicine.
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Rights and permissions
About this article
Cite this article
Ehret, G., O'Connor, A., Weder, A. et al. Follow-up of a major linkage peak on chromosome 1 reveals suggestive QTLs associated with essential hypertension: GenNet study. Eur J Hum Genet 17, 1650–1657 (2009). https://doi.org/10.1038/ejhg.2009.94
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.94
Keywords
This article is cited by
-
Immune and inflammatory mechanisms in hypertension
Nature Reviews Cardiology (2024)
-
Rat models of human diseases and related phenotypes: a systematic inventory of the causative genes
Journal of Biomedical Science (2020)
-
Immune mechanisms of salt-sensitive hypertension and renal end-organ damage
Nature Reviews Nephrology (2019)
-
Long non-coding RNA and mRNA analysis of Ang II-induced neuronal dysfunction
Molecular Biology Reports (2019)
-
Polymorphism rs7555523 in transmembrane and coiled-coil domain 1 (TMCO1) is not a risk factor for primary open angle glaucoma in a Saudi cohort
Journal of Negative Results in BioMedicine (2016)
