
Abstract
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder caused by homozygous mutations of the SMN1 gene. Three forms of SMA are recognized (type I–III) on the basis of clinical severity. All patients have at least one or more (usually 2–4) copies of a highly homologous gene (SMN2), which produces insufficient levels of functional SMN protein, because of alternative splicing of exon 7. Recently, evidence has been provided that SMN2 expression can be enhanced by pharmacological treatment. However, no reliable biomarkers are available to test the molecular efficacy of the treatments. At present, the only potential biomarker is the dosage of SMN products in peripheral blood. However, the demonstration that SMN full-length (SMN-fl) transcript levels are reduced in leukocytes of patients compared with controls remains elusive (except for type I). We have developed a novel assay based on absolute real-time PCR, which allows the quantification of SMN1-fl/SMN2-fl transcripts. For the first time, we have shown that SMN-fl levels are reduced in leukocytes of type II–III patients compared with controls. We also found that transcript levels are related to clinical severity as in type III patients SMN2-fl levels are significantly higher compared with type II and directly correlated with functional ability in type II patients and with age of onset in type III patients. Moreover, in haploidentical siblings with discordant phenotype, the less severely affected individuals showed significantly higher transcript levels. Our study shows that SMN2-fl dosage in leukocytes can be considered a reliable biomarker and can provide the rationale for SMN dosage in clinical trials.
Similar content being viewed by others


Introduction
Proximal spinal muscular atrophies (SMA) are a group of clinically variable motor neuron disorders characterized by the degeneration of the anterior horn cells of the spinal cord. On the basis of age of onset and severity of the clinical course, childhood-onset SMA can be classified into three forms (type I–III). SMA III patients can be further divided into type IIIa and IIIb on the basis of whether the onset is below or over the age of 3 years, respectively.1 SMAI-III are autosomal recessive, and are caused by loss of function of the survival motor neuron (SMN1) gene.2 SMN1 and a nearly identical copy, SMN2, are located in a duplicated inverted region at 5q13. Both genes encode the SMN protein but, because of alternative splicing, the majority of SMN2 transcripts lack exon 7 (SMN-delta7), and are unable to produce a sufficient amount of protein to prevent the onset of the disease. The SMN protein is expressed in most tissues and is localized in the cytoplasm and in the nucleus. It has been shown that the level of SMN protein is markedly reduced in SMA patients, both in spinal cord and in cell cultures and inversely correlate with phenotypic severity.3, 4, 5 Patients can carry a variable copy number of the SMN2 gene, higher copy numbers being generally associated with milder phenotypes.6, 7, 8
At present, no cure for SMA is available. Recently, evidence has been provided that SMN2 gene expression can be enhanced by pharmacological treatment in vivo and/or in vitro, using different compounds.9, 10, 11, 12, 13, 14, 15, 16, 17 The clinical efficacy of some of these compounds has been tested also in clinical trials.18, 19, 20, 21
The advances in SMA clinical research highlight the need of reliable biomarkers to monitor the efficacy at the molecular level of treatments during trials, the dosage of SMN transcripts or protein in peripheral blood samples being the only one potentially available. However, possible variations of SMN transcripts/protein levels as evaluated in leukocytes may not reflect the real effect of pharmacological treatment in target tissues, such as spinal cord and, eventually, skeletal muscle. So far, some assays have been developed and validated for SMN2 transcript14, 22, 23, 24 or protein25, 26 quantification. However, to date it has not been shown whether SMN full-length (SMN-fl) transcript or protein levels in leukocytes differ significantly among controls, carriers, and patients. In particular, a reduction of SMN-fl levels has been shown only for type I patients.14, 22 The reported SMN mRNA assays are mainly based on relative semiquantitative PCR in which transcript levels are determined by normalizing with respect to housekeeping gene transcript levels, used as endogenous controls.22, 23, 24 However, it has been shown that the expression levels of these genes vary widely in the general population and/or can be putatively affected by pharmacological treatments or metabolic status, thus reducing the sensitivity of the earlier published assays.14 Brichta et al14 have developed a real-time PCR assay based on the measurement of SMN levels relative to the amount of RNA. We have developed an alternative molecular test based on absolute real-time PCR that allows the quantification of the number of SMN1-fl and SMN2-fl mRNA molecules per nanogram of total RNA (mol/ng) and is suitable for measuring SMN transcripts, thus avoiding possible biases because of the variations in endogenous control transcript levels. In our assay, GAPDH transcripts quantification has been included as positive control for PCR amplification and to rule out that possible differences between patients and controls could be related to PCR efficiency or RNA quality (see Supplementary information). We have used this novel assay to investigate whether SMN-fl transcript levels are reduced in patients compared with controls, and to assess whether transcript levels correlate with phenotypic severity in patients, which are the prerequisites for using SMN dosage as a biomarker for future clinical trials in SMA patients.
Subjects and methods
Subjects
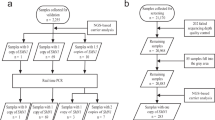
Blood samples were obtained from 51 SMA patients (2 type I, 16 type II, and 33 type III), 23 carriers, and 28 controls. The characteristics of age and sex ratio of the three groups are summarized in Table 1. All patients had homozygous absence of SMN1 exon 7. Type III SMA patients have been subgrouped according to the classification proposed by Zerres1 in type IIIa and IIIb. For type II patients between 2.5 and 12 years of age, functional ability was evaluated using the Hammersmith functional motor scale.27
Among patients, six sib pairs were analyzed: five pairs were phenotypically discordant (type IIIa sister/II brother; type IIIa/II sisters; asymptomatic/IIIb sisters; type IIIb brother/IIIa sister; oligosymptomatic/type IIIb brothers), and one further pair of sibs who were phenotypically similar (type IIIb brother and sister). Carriers were either parents of patients or were selected among relatives of patients or other relatives who tested positive for carrier status. For patients and carriers, both total RNA and genomic DNA were extracted. Controls were healthy individuals, seen at the Genetics Clinic of the Catholic University Hospital for karyotype analysis. Blood samples from controls were rendered anonymous and used for RNA extraction only. Individuals taking drugs known to modify SMN expression were excluded from our cohorts. For repetitive samplings, blood samples were drawn approximately at the same hour during the morning, to rule out possible biases because of circadian variations in SMN/GAPDH transcript levels or in feeding. Finally, four fibroblast cell cultures were analyzed in this study, one from a control and three from patients (one for each type of SMA).
DNA extraction, SMN2 gene copy number assessment, RNA extraction, and RT-PCR
The DNA, extracted by the standard salting-out procedures, was quantified by absorbance at 260 and 280 nm (GeneQuant Pro, Pharmacia Biotech, Arlington Heights, IL, USA). SMN2 gene copy number, as well as carrier status, was determined as reported earlier.12 For RNA extraction from peripheral blood, PAXgene blood RNA tubes (BD Biosciences, San Jose, CA, USA) and kit (Qiagen) were used. In the case of fibroblast cultures, total RNA was extracted by RNeasy mini kit (Qiagen, Duesseldorf, Germany). For all RNA samples, concentration was established by absorbance determination and quality was assessed by agarose gel electrophoresis. A total of 2 μg of total RNA were used for RT-PCR using a High Capacity cDNA Archive Kit (Applied Biosystems, Carlsbad, CA, USA) in a final reaction volume of 25 μl, using random primers for reverse transcription.
External standard constructs design
Three plasmids were constructed for SMN1-fl, SMN2-fl, and GAPDH, respectively, by amplifying a control cDNA. SMN1 and SMN2 were amplified by using the primer pair: SMN_exst-F: 5′-GCTTTGGGAAGTATGTTAATTTCA-3′ and SMN_exst-R: 5′-CTATGCCAGCATTTCTCCTTAATT-3′, located in exon 6 and exon 7/8 junction, respectively. For GAPDH, primer pair GAPDH_exst-F: 5′-CTCTGCTGATGCCCCCATGTTCGT-3′ and GAPDH_exst-R: 5′-CAAAGTTGTCATGGATGACCTTGG-3′, located in exon 5 and exon 6, respectively, was used. For SMN1/SMN2 and GAPDH genes, PCR products of 129 and 133 bp, respectively, were obtained. Subsequently, PCR products were cloned by using TA cloning kit (Qiagen). Plasmid DNA was extracted by the QIAprep Spin Miniprep Kit (Qiagen) and quantified both by absorbance and by agarose gel electrophoresis with scaling serial dilution of lambda DNA. The presence of possible sequence variations in plasmids, randomly introduced by Taq polymerase, was ruled out by sequence analysis of the clones, performed using the ABI-Prism 3130 instrument and BigDye terminator v3.1 Cycle Sequencing kit (Applied Biosystems). On the basis of plasmid length (3980 and 3984 bp for SMN1/SMN2 and GAPDH, respectively), the molecular weight and the number of plasmid molecules per nanogram of DNA (around 2.29 × 108/ng for the three plasmids) were determined. Single use serial dilutions of the three external standards, ranging from 104 to 107 molecules, were aliquoted and kept frozen at −80°C.
Primers and MGB-probes
Primer Express v1.5 software (Applied Biosystems) was used to design optimized minor groove binder (MGB) probes and primers for real-time RT-PCR. SMN1 and SMN2-fl transcripts were amplified by using the same primer pair (SMN_abs-F: 5′-TACATGAGTGGCTATCATACTGGCTA-3′ and SMN_abs-R: 5′-AATGTGAGCACCTTCCTTCTTTTT-3′, located in exons 6 and 7, respectively), obtaining 72 bp PCR products. Full-length transcripts of the two genes were specifically distinguished by two different Taqman MGB probes, labeled with different fluorochromes, on the basis of the C-T transition located in exon 7 (SMN1_abs: 5′-NED-TATGGGTTTCAGACAAA-NFQ-3′ and SMN2_abs: 5′-VIC-ATATGGGTTTTAGACAAAA-NFQ-3′). For GAPDH, an amplicon of 73 bp was obtained by using the primer pair GAPDH_abs-F: 5′-GGGTGTGAACCATGAGAAGTATGA-3′ and GAPDH_abs-R: 5′-CTAAGCAGTTGGTGGTGCAGG-3′. MGB probe sequence was: 5′-FAM-CAAGATCATCAGCAATGC-NFQ-3′.
Real-time PCR assay and construction of standard curves
The PCR reactions were performed in a final volume of 20 μl, containing 2 × Taqman Universal Mastermix (Applied Biosystems), 40 ng of cDNA (or appropriate dilutions of external standards), appropriate concentrations of SMN primers, SMN1 and SMN2 probes, or of GAPDH primers and probe. Each sample was amplified in quadruplicate and each experiment repeated at least twice. All reactions were performed using 7900HT Fast Real-Time PCR System (Applied Biosystems). Optimal primer and probe concentrations were as follows: SMN_abs_F and SMN_abs_R: 900 nM; GAPDH_abs_F and GAPDH_abs_R: 30 nM; SMN1_abs, SMN2_abs, GAPDH_abs: 200 nM. Serial dilutions of external standards, ranging from 104 to 107 copies were used to construct the standard curves. We did not use plasmid concentrations lower than 104 copies because of the poor stability of such dilutions. The number of SMN1-fl, SMN2-fl, and GAPDH mRNA molecules was extrapolated automatically by the Sequence Detection System v2.2.2 software (Applied Biosystems).
Statistical analysis
Statistical analysis was performed by using Statgraphics-Centurion XV.II (Statpoint Technologies, Warrenton, VA, USA) software. The experimental variability and the reproducibility of the real-time PCR assay were assessed by determining the mean and standard deviation (SD) of coefficient of variation (CV) of repeated experiments. For each sample, the CV was determined as the ratio between the SD and mean transcript levels of repeated amplifications. The distribution of SMN1-fl, SMN2-fl, total SMN-fl (SMN1-fl plus SMN2-fl), and GAPDH transcripts was analyzed by using Kolmogorov–Smirnov, Shapiro–Wilks’ W, and Lilliefors tests for normality. Possible alternative distributions of SMN transcripts were also evaluated by using goodness-of-fit tests.
To compare transcript levels in the three populations, both parametric (t-test for independent variables and one-way ANOVA) and non-parametric tests (Kruskal–Wallis ANOVA by ranks (KW) and Mann–Whitney U-test (MW)) were used. Possible correlations between SMN1-fl and SMN2-fl transcript levels, between SMN2 gene copy number and SMN2-fl transcript levels, as well as between Hammersmith's functional motor scale score and SMN2-fl transcript levels were analyzed by a linear regression model. Contingency tables and two-tailed Fisher's F-test were used to calculate the relative risk (RR) and 95% confidence interval (CI) for correlations of age of onset and SMN2-fl transcript levels. To evaluate possible differences in SMN2-fl levels in siblings, the hypothesis test was used: pairs were divided on the basis of phenotype and the relatively less severe sibs were compared with more severe ones. For all tests, significance cutoff was fixed at P-values ≤0.05.
Results
Validation, specificity, and reproducibility of the assay are described in Supplementary information. The exact number of SMN2 genes, SMN-fl, SMN1-fl, SMN2-fl, and GAPDH transcript levels of single individuals are indicated in Supplementary Table 1.
SMN-fl transcripts do not show a normal distribution
The normality tests reported in the Subjects and methods section indicated that in carriers and controls, SMN1-fl, SMN2-fl, and SMN-fl levels do not show a normal distribution (P <0.05, Supplementary Figure 1a-b and data not shown). Similar results were obtained for SMN2-fl levels in patients (P <0.04, Supplementary Figure 1c and Table 1). Therefore, we considered median, quartiles, minimum and maximum as more appropriate to describe SMN levels than mean±SD.
SMN2-fl levels are more stable over time than SMN1-fl
To evaluate physiological fluctuations of SMN1-fl, SMN2-fl, and GAPDH transcripts, we performed 2–4 blood samplings in seven controls during a period of 1 month (at days 0, 1, 14, and 30) and two blood draws in six patients (at day 0 and 30). The results are summarized in Figure 1. Although a certain degree of fluctuation was observed, SMN2-fl and GAPDH transcript levels seemed to be more stable over time than SMN1-fl. SMN2-fl levels seemed to be less variable in SMA patients than in controls. To confirm this observation, we have evaluated the mean CV (0.19±0.11 and 0.14±0.06 for SMN2-fl and GAPDH, respectively), which was more similar to that expected for experimental variation of the assay (see Assay development and validation section in online Supplementary information). In contrast, SMN1-fl transcripts showed wide day-to-day variations (mean CV: 0.35±0.17). We also evaluated the total SMN-fl level variations and observed that transcript level fluctuations reflect that observed for SMN1-fl levels (data not shown). As SMN1-fl and SMN2-fl transcripts are amplified by the same primer pair, the different mean CV of the two amplicons cannot be ascribed to the PCR artifact.

(a) Day-to-day variations of SMN-fl, SMN1-fl, and SMN2-fl transcript levels in seven controls (ctrl 1–7) and of SMN2-fl 2 type III patients (pt 1–6). SMN2-fl level fluctuations were similar to that expected for experimental variability, whereas in controls SMN1-fl levels and, consequently, SMN-fl levels varied up to threefold. (b) GAPDH transcript level fluctuations were in the range of experimental variability.
Patients have lower SMN levels compared with controls and carriers
To assess whether SMN-fl transcripts are reduced in SMA subjects, we compared SMN2-fl levels in patients (n=51) with SMN-fl in controls (n=28, Table 1 and Figure 2). The difference between the two groups was statistically significant (MW: P=4.3 × 10−5, KW: P=4.2 × 10−5); also, when excluding the two type I patients, the P-values remained highly significant (MW=KW, P=8 × 10−5). We subsequently subdivided the patients according to their SMA type. Although the number of samples from type I patients (n=2) was insufficient for statistical analysis, the difference in transcript levels between patients and controls was statistically significant both for SMA type II (n=16, MW: 2.4 × 10−5, KW: P=2.22 × 10−5) and type III (n=33, MW and KW: P=0.0042). No significant differences in SMN2-fl levels were observed when dividing patients by sex (MW and KW: P=0.16) or age (< or ≥14 years, MW: P=0.07, KW: P=0.06).

SMN2-fl levels in patients vs SMN-fl in controls. SMN2-fl transcript levels in patients are significantly reduced compared with SMN-fl levels in controls. Also median SMN2-fl levels in patients are significantly lower compared with controls when considering type II and III patients separately. Type II patients showed significantly lower SMN2-fl transcript levels compared with type III subjects.
SMN2-fl levels are not related to SMN2 gene copy number
To evaluate whether SMN2-fl levels correlate with SMN2 gene copy number, we have determined the gene copies in 35 of 51 patients. The two SMA type I patients had two copies, 25 SMA type II and III patients had three copies, and eight type III patients had four copies. By using a linear correlation model, indicating SMN2 copy number as an independent variable, no evidence for a correlation between SMN2-fl levels and gene copy number was found (one-way ANOVA P=0.52). Moreover, when comparing patients with three and four SMN2 genes (Figure 3), no statistical difference was found (MW=KW: P=0.35), although individuals with four copies had slightly higher median SMN2-fl transcript levels (3 SMN2: 56.25 mol/ng, range: 26.75–121.5; 4 SMN2: 63.75 mol/ng, range: 41.25–112.25).

SMN2-fl transcript levels are not related to SMN2 gene copy number. Although patients with four SMN2 copies showed higher median SMN2-fl levels compared with individuals with three SMN2, this difference was not statistically significant.
SMN2-fl transcript levels are related to phenotypic severity
To assess whether SMN2-fl levels are related to clinical severity, different parameters were evaluated: type of SMA, age at onset, and the Hammersmith motor scale score (in type II patients with age range 2.5–12 years). Type II patients showed median SMN2-fl transcript levels lower than type III, and the difference between the two groups was highly significant (MW=KW: P=0.0034, Table 1 and Figure 2). Subsequently, SMN2-fl transcript levels were related to the age of onset of type II and III patients. Although in type II patients no correlation was found between the two variables (data not shown), in the case of SMA type III (n=23, 1.5–19 years) age of onset was inversely related to SMN2-fl levels. Although it was not possible to identify a linear regression model between the two variables, we found that SMN2-fl levels ≥58 mol/ng are related to a threefold lower risk of disease onset before the age of 3 (RR: 0.31, CI: 0.12–0.76, Fisher's exact test P=0.02, Figure 4a). A total of 6 out of 12 type IIIb and 2 out of 10 type IIIa patients had four SMN2 copies. The other patients had three SMN2 genes.

Correlation between SMN2-fl levels and clinical severity (a) Transcript levels are related to the age of onset of type III patients: patients with SMN2-fl levels <58 mol/ng of total RNA showed a threefold higher risk of disease onset below the age of 3 years (type IIIa). (b) In type II patients (ranging: 2.5–12 years), a linear correlation was found between SMN2-fl levels and Hammersmith functional scale scores. Dots indicate SMN2-fl levels and the corresponding functional score for individual patients; the black line is the graphic representation of the equation describing the linear regression model; the dark gray lines indicate 95% confidence interval and the outer light gray lines are 95% prediction limits for new observations. (c) Patients with a functional score ≤20 showed significantly lower SMN2-fl levels. (d) In haploidentical sib pairs with discordant SMA phenotype, the less severely affected sib (white columns) showed significantly higher transcript levels compared with the more severely affected one (black columns), whereas phenotypically concordant sibs (gray columns) had similar SMN2-fl levels. However, SMN2nn-fl transcript quantification is not predictive of phenotypic severity in individual cases, as less severely affected patients may have higher transcript levels. Error bars indicate SD of repeated experiments
The motor ability of type II patients was evaluated by the Hammersmith scale score.27 The cutoff of 12 years was chosen to avoid bias because of the presence of complications, such as severe scoliosis and contractures, which are more frequent after this age. Only 10 out of 16 type II patients were below the age of 12 years. We compared SMN2-fl levels and motor function by using a linear regression model, indicating transcript levels as an independent variable, and found a significant correlation between the two variables (β=0.64, P=0.04, Figure 4b), indicating a moderately strong association between the Hammersmith score and transcript levels. The relationship between the two variables can be described by using the following expression:

Subsequently, we subgrouped patients on the basis of functional scores ≤20 (n=5) or >20 (n=5), and observed that the more severely affected patients had median lower SMN2-fl levels (29.25 mol/ng, range: 23.60–33.50), compared with the other group (median: 57.00 mol/ng, range: 45.5–102.75); the difference was statistically significant (MW: P=0.01, KW: P=0.009, Figure 4c).
Finally, to assess whether differences in SMN2-fl levels exist between haploidentical SMA siblings, we evaluated transcript levels in six sib pairs, five of which showed marked phenotypic differences, whereas one further sib pair was similar in disease severity. We found that in discordant couples, the less severely affected sib showed significantly higher SMN2-fl levels compared with the more severely affected one (P=0.002). The phenotypically similar sib pair showed only slightly different SMN2-fl levels (Figure 4d).
Discussion
The recent move in SMA research from basic to clinical has raised the necessity to develop reliable and reproducible clinical tools, and to identify biomarkers useful for monitoring the response of SMA patients to pharmacological treatments. Although validated clinical tools have been developed, at present the quantification of SMN2 gene products in blood leukocytes, either at protein or transcript levels, is the only potential biomarker available. However, except for type I SMA,14, 22 no clear differences in SMN levels between patients and controls have been shown, thus questioning the reliability of transcript analysis as a biomarker for SMA and its usefulness in monitoring the molecular effects of pharmacological treatment. We have developed and validated a new real-time PCR assay, based on the use of absolute standard curves, which allows the quantification of SMN-fl transcripts as the number of mRNA molecules per nanogram of total RNA, independently from the use of endogenous controls. We have exploited techniques that are widely used to determine the plasmatic load of some RNA viruses, such as HIV28 or HCV,29 or of prion protein.30
One interesting finding of our study is the difference in day-to-day variability of SMN1-fl and SMN2-fl levels. Although we have observed moderate variations in SMN2-fl levels, which were similar to that expected for experimental variability and comparable with that of GAPDH transcripts, SMN1-fl levels varied up to threefold from one day to the other. Owing to SMN1-fl variability, the total amount of SMN-fl transcripts also varied markedly (Figure 1a and b and data not shown; see also Supplementary information). The relevance of this observation is related to the strategy to test the in vivo effect of different compounds on SMN expression by administering a given drug to parents of SMA patients, who often offer spontaneously their own collaboration.13, 14 In a study on the molecular effect of valproic acid, 7 of 10 carriers had increased levels of SMN-fl transcripts after treatment, whereas only in one-third of the patients a molecular effect was shown.14 The discrepancy between these observations may be explained by spontaneous fluctuations in SMN1-fl transcript levels, as the assay used by these authors does not allow the discrimination of SMN1 from SMN2 transcripts. Thus, the results of molecular studies of the in vivo efficacy of a given compound on SMN expression in carriers or controls should be interpreted cautiously, and only data relative to SMN2 transcripts should be taken into account.
The most relevant finding of our study is that SMN2-fl transcripts are significantly reduced in type II and III patients. As indicated in Figure 2 and the Table 1, both considering all patients as a group or divided on the basis of the type of SMA, SMN2-fl transcripts are significantly reduced in the majority of SMA patients, compared with SMN-fl in controls. Moreover, type III patients have significantly higher SMN2-fl transcript levels than type II. To our knowledge, this is the first demonstration of a statistically significant reduction of SMN levels in blood leukocytes of type II–III patients. The partial overlap of SMN-fl levels between patients and controls is not unexpected: blood leukocytes are not target cells in SMA, and it is conceivable that in target tissues, the cutoff between patients and controls could be sharper. It would be important to study the key cell types involved in SMA pathophysiology, such as motor neurons and/or muscle cells and to relate SMN2-fl levels in these cells with those found in blood.
To gain further insights into a possible correlation between transcript levels and clinical severity, we investigated whether SMN2-fl levels are related to the age of onset of type II and III patients, and to the Hammersmith motor scale score (for type II patients below the age of 12 years). We did not observe any correlation between age of onset of type II patients and SMN2-fl transcript levels; however, in these patients, the first symptoms of the disease are often misrecognized by parents and may be interpreted as a slight delay in ambulation achievement. In the case of type III patients, we observed that higher transcript levels are related to more advanced age at onset (Figure 4a). In particular, we found that patients with SMN2-fl levels ≥58 mol/ng have a threefold lower risk of disease onset below the age of 3 years. The onset of the disease below or over this age (type IIIa and b) was earlier indicated as an important prognostic factor for walking ability maintenance:1 in an earlier study, Wirth et al8 reported that the 60% of type IIIb and 35% of type IIIa patients have four SMN2 copies (50 and 20%, respectively, in our cohort), confirming the correlation between phenotypic severity and SMN2 gene number. A correlation between clinical severity and SMN2-fl transcript levels is further supported by the finding that type II children with higher scores of the Hammersmith functional motor scale show significantly higher SMN2-fl transcript levels compared with those with lower scores (Figure 4b-c). In addition, in haploidentical SMA siblings with discordant phenotype, the less severely affected sib showed significantly higher SMN2-fl levels compared with the more severely affected one, whereas in a phenotypically similar pair, both sibs had similar SMN2-fl levels (Figure 4d).
We and others have earlier found a correlation between SMN2 copy number and phenotypic severity;6, 7, 8 however, SMN2 gene number alone is not sufficient to explain the phenotypic variability of SMA, as patients with the same gene number have different phenotypes, and haploidentical sibs can be markedly discordant for disease severity. To our knowledge, this is the first in vivo molecular study on SMA discordant siblings; only one in vitro study has been published earlier,31 showing that the more severely affected sibs had lower SMN levels in lymphoblastoid cell lines but not in fibroblasts. We compared the SMN2-fl transcript levels and SMN2 gene number and did not find a significant correlation (Figure 3). Similar results were also found in the study by Simard et al,23 whereas Sumner et al22 and Vezain et al24 found that SMN2 transcript levels are related to the number of SMN2 genes. The discrepancy among different studies can be at least partially accounted for by the use of relative quantification and of different endogenous internal standards that vary widely among different individuals. Our data suggest that the regulation of SMN2-fl transcript production (at transcriptional or splicing levels) has greater influence on phenotypic modulation than SMN2 copy number per se, which is further supported by the finding that in discordant sib pairs with identical gene copy number, the less severely affected sib higher transcript levels. However, although SMN2-fl transcript quantification appears to be more tightly related to phenotypic variability than SMN2 copy number assessment, it should not be used as a prognostic marker for individual patients, because of the overlapping between the different phenotypic groups. It would be of interest to have further data on a possible correlation between SMN transcript and protein levels in leukocytes by means of an ELISA assay, which is currently available for cell cultures only.26
In conclusion, our data indicate that SMN2-fl quantification in blood leukocytes by absolute real-time PCR can be considered suitable as a biomarker in SMA clinical trials as SMN2-fl transcripts are both reduced in patients compared with SMN-fl in controls and their level may at least partially reflect SMN levels in target tissues, being related to disease severity. The present assay, compared with that published earlier, offers the opportunity to standardize and optimize SMN2-fl transcript quantification in different laboratories, in the view of upcoming international multicentric clinical trials. Further studies are still necessary to assess whether possible SMN2-fl transcript increments may correlate with clinical improvements of SMA patients included in clinical trials, and/or if SMN2-fl dosage can predict clinical response to pharmacological treatment.
References
Zerres K, Rudnik-Schoneborn S, Forrest E et al: A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci 1997; 146: 67–72.
Lefebvre S, Bürglen L, Reboullet S et al: Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995; 80: 155–165.
Lefebvre S, Burlet P, Liu Q et al: Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 1997; 16: 265–269.
Coovert DD, Le TT, McAndrew PE et al: The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 1997; 6: 1205–1214.
Patrizi AL, Tiziano F, Zappata S : SMN protein analysis in fibroblast, amniocyte, and CVS cultures from spinal muscular atrophy patients and its relevance for diagnosis. Eur J Hum Genet 1999; 7: 301–309.
Feldkötter M, Schwarzer V, Wirth R et al: Quantitative analysis of SMN1 and SMN2 based on real-time LightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002; 70: 358–368.
Tiziano FD, Bertini E, Messina S et al: The Hammersmith functional score correlates with the SMN2 copy number: a multicentric study. Neuromuscul Disord 2007; 17: 400–403.
Wirth B, Brichta L, Schrank B et al: Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet 2006; 119: 422–428.
Chang J-G, Hsieh-Li H-M, Jong Y-J et al: Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci USA 2001; 98: 9808–9813.
Brichta L, Hofmann Y, Hahnen E et al: Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet 2003; 12: 2481–2489.
Sumner CJ, Huynh TN, Markowitz JA et al: Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann Neurol 2003; 54: 647–654.
Andreassi C, Angelozzi C, Tiziano FD et al: Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet 2004; 12: 59–65.
Brahe C, Vitali T, Tiziano FD et al: Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. Eur J Hum Genet 2005; 13: 256–259.
Brichta L, Holker I, Haug K : In vivo activation of SMN in spinal muscular atrophy carriers and patients treated with valproate. Ann Neurol 2006; 59: 970–975.
Grzeschik SM, Ganta M, Prior TW et al: Hydroxyurea enhances SMN2 gene expression in spinal muscular atrophy cells. Ann Neurol 2005; 58: 194–202.
Jarecki J, Chen X, Bernardino A et al: Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy. Hum Mol Genet 2005; 14: 2003–2018.
Angelozzi C, Borgo F, Tiziano FD : Salbutamol increases SMN mRNA and protein levels in spinal muscular atrophy cells. J Med Genet 2008; 45: 29–31.
Pane M, Staccioli S, Messina S et al: Daily salbutamol in young patients with SMA type II. Neuromuscul Disord 2008; 18: 536–540.
Mercuri E, Bertini E, Messina S et al: Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology 2007; 68: 51–55.
Weihl CC, Connolly AM, Pestronk A : Valproate may improve strength and function in patients with type III/IV spinal muscle atrophy. Neurology 2006; 67: 500–501.
Liang WC, Yuo CY, Chang JG et al: The effect of hydroxyurea in spinal muscular atrophy cells and patients. J Neurol Sci 2008; 268: 87–94.
Sumner CJ, Kolb SJ, Harmison GG et al: SMN mRNA and protein levels in peripheral blood. Neurology 2006; 66: 1067–1073.
Simard LR, Bélanger M-C, Morissette S et al: Preclinical validation of a multiplex real-time assay to quantify SMN mRNA in patients with SMA. Neurology 2007; 68: 451–456.
Vezain M, Saugier-Veber P, Melki J et al: A sensitive assay for measuring SMN mRNA levels in peripheral blood and in muscle samples of patients affected with spinal muscular atrophy. Eur J Hum Genet 2007; 15: 1054–1062.
Kolb SJ, Gubitz AK, Olszewski Jr RF et al: A novel cell immunoassay to measure survival of motor neurons protein in blood cells. BMC Neurol 2006; 6: 6.
Thi Man N, Humphrey E, Lam LT et al: A two-site ELISA can quantify upregulation of SMN protein by drugs for spinal muscular atrophy. Neurology 2008. epub Fpage: ahead Misc: of print.
Main M, Kairon H, Mercuri E et al: The Hammersmith functional motor scale for children with spinal muscular atrophy: a scale to test ability and monitor progress in children with limited ambulation. Eur J Paediatr Neurol 2003; 7: 155–159.
Gueudin M, Simon F : Plasma RNA viral load in HIV-1 group O infection by real-time PCR. Methods Mol Biol 2005; 304: 221–228.
Castelain S, Descamps V, Thibault V et al: TaqMan amplification system with an internal positive control for HCV RNA quantitation. J Clin Virol 2004; 31: 227–234.
Tichopad A, Pfaffl MW, Didier A : Tissue-specific expression pattern of bovine prion gene: quantification using real-time RT-PCR. Mol Cell Probes 2003; 17: 5–10.
Helmken C, Hofmann Y, Schoenen F et al: Evidence for a modifying pathway in SMA discordant families: reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum Genet 2003; 114: 11–21.
Acknowledgements
This study has been granted by ASAMSI, Famiglie SMA, and FSMA USA. We are grateful to the patients and their families for participating in the study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Tiziano, F., Pinto, A., Fiori, S. et al. SMN transcript levels in leukocytes of SMA patients determined by absolute real-time PCR. Eur J Hum Genet 18, 52–58 (2010). https://doi.org/10.1038/ejhg.2009.116
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.116
Keywords
This article is cited by
-
Biomarkers in 5q-associated spinal muscular atrophy—a narrative review
Journal of Neurology (2023)
-
Spinal Muscular Atrophy (SMA) in the Therapeutic Era
Current Genetic Medicine Reports (2019)
-
A rare variant (c.863G>T) in exon 7 of SMN1 disrupts mRNA splicing and is responsible for spinal muscular atrophy
European Journal of Human Genetics (2016)
-
Decay in survival motor neuron and plastin 3 levels during differentiation of iPSC-derived human motor neurons
Scientific Reports (2015)
-
Clinical and molecular cross-sectional study of a cohort of adult type III spinal muscular atrophy patients: clues from a biomarker study
European Journal of Human Genetics (2013)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}