
Abstract
Cardio-facio-cutaneous syndrome (CFCS) is a rare disease characterized by mental retardation, facial dysmorphisms, ectodermal abnormalities, heart defects and developmental delay. CFCS is genetically heterogeneous and mutations in the KRAS, BRAF, MAP2K1 (MEK1) and MAP2K2 (MEK2) genes, encoding for components of the RAS–mitogen activated protein kinase (MAPK) signaling pathway, have been identified in up to 90% of cases. Here we screened a cohort of 33 individuals with CFCS for MEK1 and MEK2 gene mutations to further explore their molecular spectrum in this disorder, and to analyze genotype–phenotype correlations. Three MEK1 and two MEK2 mutations were detected in six patients. Two missense MEK1 (L42F and Y130H) changes and one in-frame MEK2 (K63_E66del) deletion had not been reported earlier. All mutations were localized within exon 2 or 3. Together with the available records, the present data document that MEK1 mutations are relatively more frequent than those in MEK2, with exons 2 and 3 being mutational hot spots in both genes. Mutational analysis of the affected MEK1 and MEK2 exons did not reveal occurrence of mutations among 75 patients with Noonan syndrome, confirming the low prevalence of MEK gene defects in this disorder. Clinical review of known individuals with MEK1/MEK2 mutations suggests that these patients show dysmorphic features, ectodermal abnormalities and cognitive deficit similar to what was observed in BRAF-mutated patients and in the general CFCS population. Conversely, congenital heart defects, particularly mitral valve and septal defects, and ocular anomalies seem to be less frequent among MEK1/MEK2 mutation-positive patients.
Similar content being viewed by others

Introduction
Cardio-facio-cutaneous syndrome (CFCS) is a sporadic multiple congenital anomaly/mental retardation disorder characterized by failure to thrive, severe feeding problems, developmental delay, reduced growth, distinctive dysmorphic face, ectodermal abnormalities and heart defects.1 CFCS is genetically heterogeneous, with mutations in the KRAS, BRAF, MAP2K1 (MEK1) and MAP2K2 (MEK2) genes occurring in approximately 62.5–90% of affected individuals.2, 3, 4, 5 CFCS disease genes encode for signal transducers participating in the RAS–MAPK pathway, which is known to play a crucial role in development and oncogenesis.6 Remarkably, dysregulated function of these and other proteins implicated in this signaling cascade has been recently shown to represent the molecular cause underlying a group of disorders clinically related to CFCS, including Noonan syndrome (NS), LEOPARD syndrome and Costello syndrome.5, 7, 8, 9, 10, 11, 12, 13, 14 On the basis of the shared pathogenic mechanism and clinical overlap, these disorders are now grouped in the recently coined neuro-CFCS family.15
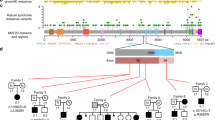
In approximately 20% of cases, CFCS is caused by mutations affecting the MEK1 and MEK2 genes.3, 4, 16, 17, 18 MEK1 and MEK2 are dual-specificity protein kinases, which work as effectors of the serine/threonine kinase RAF family members by phosphorylating and activating ERK proteins.19 MEK proteins are composed of a regulatory domain at the N-terminus, a single kinase domain and a C-terminal tail (Figure 1). Available mutation data support the view that the vast majority of CFCS-causing mutations are missense changes affecting residues located in the regulatory region and the N-terminal portion of the catalytic domain. Until now, mutations in the MEK1 and MEK2 genes have been detected in 58 patients with CFCS, and in three patients exhibiting a phenotype apparently fitting NS.16 Although these findings would be suggestive of phenotypic heterogeneity, the clinical features associated with mutations in the MEK1 and MEK2 genes have not been delineated exhaustively. The presence of normal cognitive abilities in a minor percentage of individuals seems to suggest that MEK1/MEK2 mutation-positive patients show a milder clinical phenotype than what is observed in patients with BRAF mutations or in the general CFCS population.

MEK1 and MEK2 mutations in cardio-facio-cutaneous syndrome. Genomic characterization of the MEK1 and MEK2 genes. Exons are represented by bars and numbered. Orange boxes indicate regions coding for the protein kinase domain (residues 68–361 in MEK1 and 72–369 in MEK2). Earlier identified mutations are represented as blue squares,2, 3, 4, 16, 17, 18, 20, 21 whereas those identified in this study are colored yellow.
To further characterize the molecular spectrum of MEK1 and MEK2 gene mutations and the phenotypic features associated with these molecular defects, we screened cohorts including patients with CFCS and NS, and carried out a detailed clinical characterization of mutation-positive cases. To explore possible genotype–phenotype correlations, we also compared the phenotype of MEK1/MEK2 mutation-positive CFCS cases with those associated with BRAF mutations, as well as with the general CFCS population.
Material and methods
Patients
Two cohorts were included in the study. A first group included 33 sporadic patients with a full-blown CFCS phenotype. Clinical diagnosis of CFCS was based on the presence of clinical features such as craniofacial dysmorphisms, cardiac anomalies, characteristic ectodermal and musculoskeletal anomalies, postnatal growth deficiency, hypotonia and developmental delay.1 A second group comprised 75 patients with NS or exhibiting a phenotype that was suggestive for the disorder. Informed consent was obtained for all the patients included in the survey. Mutations in the PTPN11, SOS1 and RAF1 genes had been earlier excluded by denaturing high-performance liquid chromatography (DHPLC) analysis and by direct sequencing of whole coding regions in all cases. Although no KRAS mutations have been detected, 15 missense BRAF mutations were identified in 17 CFCS patients (17/33) (our unpublished data).
Mutation analysis
The patient's genomic DNA was extracted from circulating leukocytes according to the standard procedures. Mutation screening of the entire MEK1 and MEK2 coding regions and of the flanking intron–exon boundaries (CFCS cohort), or MEK1 exons 2, 3 and 6, and MEK2 exons 2, 3 and 7 (NS cohort), was carried out by DHPLC analysis (3100 WAVE DNA fragment analysis system, Transgenomic). Primer pairs, PCR and DHPLC analysis settings are available upon request. PCR products showing an abnormal DHPLC profile were reamplified and processed through PCR purification columns (Qiagen, Hilden, Germany) and sequenced using the ABI BigDye Terminator Sequencing Kit v.1.1 (Applied Biosystems, Foster City, CA).
Results
Mutation analysis
The molecular analysis identified five mutations in 6 out of 33 patients with CFCS, whereas no MEK1 or MEK2 gene variant was observed in the NS cohort (Figure 1, Table 1). Among the identified mutations, three missense changes, including the novel L42F and Y130H amino acid substitutions, affected MEK1 and occurred in four patients. One missense change and an earlier unreported short in-frame deletion (K63_E66del) in MEK2 gene were identified in two patients. Parental DNAs were available for five of the six sporadic cases, and genotyping confirmed the de novo origin of each mutation (Table 1).
MEK1 and MEK2 mutation-associated features
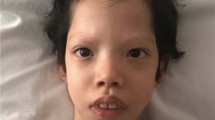
All patients with a mutated MEK1 or MEK2 allele exhibited the full-blown phenotype of the disorder (Table 2).1 Pregnancies were complicated by polyhydramnios in four cases. Weight at birth was over the 97th percentile in two of them. Recurrent facial features included macrocephaly, downslanting palpebral fissures, hypertelorism, epicanthal folds, palpebral ptosis, low set ears with thickening of the helix, flat nasal bridge, thick lips and macrostomia (Table 2, Figure 2). Ectodermal anomalies were detected in all these patients, with sparse eyebrows (6/6), sparse and/or curly hair (4/6) and keratosis pilaris (2/6) occurring most commonly. Similarly, short stature was a common feature (4/6), but the bone age was delayed in only one patient. Cardiac defects were observed in four cases, with pulmonary valve stenosis documented in three patients, and mild hypertrophic cardiomyopathy occurring in one patient. Tetralogy of Fallot with abnormal pulmonary venous return was diagnosed in one patient. Ocular abnormalities, in particular strabismus and cataract, were observed in single patients. All patients showed a variable degree of mental retardation, with the exception of patient no. 4, who was evaluated in the neonatal period (Table 2).

Dysmorphic features of subjects heterozygous for a MEK1 or MEK2 mutation. Pictures of cases N.2 (a and c) and N.6 (b and d). Note the thin and sparse hair (b), curly hair (a and c), absence of eyebrows (a, b and c), hyperkeratosis and wrinkled palm with wide squared tips (d).
Discussion
Consistent with the available records,2, 3, 4, 16, 17, 18, 20, 21, 22 these data indicate that MEK1 and MEK2 gene mutations underlie a significant fraction of CFCS, accounting for approximately 20% of cases. Of note, MEK1 mutations seem to recur more frequently compared with MEK2 defects (observed ratio 2:1), but the molecular basis for such a differential prevalence of mutations requires further investigation. Although the majority of mutations are missense changes, ours and earlier data indicate that short in-frame deletions occur in approximately 5% of cases. In our cohort, all mutations were found to affect exons 2 and 3 of both genes, confirming earlier observations indicating that the regulatory region and the adjacent N-terminal portion of the catalytic domain are mutation hot spots, possibly because of the specific perturbing effect of mutations on the autoinhibitory mechanism controlling protein activation. Nevertheless, based on the identification of rare mutations outside this region (E203Q in MEK118 and K273R in MEK23), MEK1 and MEK2 mutation screening should not be limited to these exons. Our data also confirmed that mutations affecting the tyrosine residue at codon 130 of MEK1 represent the most common event underlying CFCS, and that mutations involving residue F57 are the most frequent lesions occurring in MEK2. The perturbing role of mutations affecting these residues has been shown.4 Of note, mutations affecting the corresponding residues in the paralogs (F53 in MEK1 and Y134 in MEK2) also recur in CFCS, further supporting the relevance of these amino acids in the modulation of protein function. The novel C-to-T transition at position c.196 (L42F) in MEK1 and the in-frame c.186_197 (K63_E66 deletion) in MEK2 involve residues not found to be mutated so far. Two lines of evidence support their pathogenicity. Firstly, their de novo origin was shown for both the mutations. Secondly, multiple mutations have been documented to affect adjacent and equivalent residues in both the proteins.
To delineate more precisely the phenotypic features of patients with MEK1 and MEK2 mutations and to explore possible genotype–phenotype correlations, the clinical data of the present cohort were compared with those referred to patients with MEK or BRAF gene mutations recently provided by Armour and Allanson [21] and the CFCS cohort originally described by Kavamura et al23. From the analysis it is apparent that MEK1 and MEK2 mutation-positive patients from the present cohort show a ‘classical’ CFCS phenotype, particularly regarding presence of dysmorphisms, short stature, ectodermal features and cognitive deficits (Table 2, Figure 2). We also observed that patients with MEK and BRAF mutations share most of the clinical features, although some differences are recognizable. Specifically, downslanting palpebral fissures is more characteristic of patients with MEK mutation than of BRAF-positive patients (P<0.001). Conversely, ocular abnormalities and the presence of naevi are less common than in the BRAF-positive cohort (P<0.001) (Table 2). All of our patients and 77% of all MEK-positive patients show hypertelorism, a figure similar to that observed in BRAF-mutated patients (71%), but significantly higher (P<0.01) from the prevalence observed in the Kavamura's23 cohort (46%). These results are in disagreement with earlier studies pointing to a lower prevalence of hypertelorism among MEK1/MEK2 mutation-positive patients (73%) than of those with BRAF mutation (91%).16 Cryptorchidism has been described in the five male patients heterozygous for a MEK1/MEK2 mutation, although additional observations are necessary to support this association.
Nava et al16 suggested that patients heterozygous for a MEK1/MEK2 mutation show a milder phenotype than those with a BRAF mutation who display a higher frequency of mental retardation. In contrast, all MEK1/MEK2 mutation-positive patients in this cohort and 93% of all MEK1 or MEK2 mutation-positive patients exhibited developmental delay, which is similar to that observed among BRAF mutation-positive patients (Table 2).
Congenital heart defects (CHDs) were slightly less common in patients with mutated MEK1 or MEK2 (64%) compared with those with a mutation in BRAF (72%),22 confirming the data reported by Nava et al.16 Of note, mitral valve and septal defects seem less frequent in patients with MEK1/MEK2 mutations than in those with BRAF mutations (Table 2). Interestingly, one patient with MEK1 mutation had tetralogy of Fallot with abnormal pulmonary venous return. Tetralogy of Fallot is included among CHDs reported in clinical series of patients with NS,24 although molecular studies have documented this CHD only in cases with RAF1 mutations.10, 11 To our knowledge, the association of tetralogy of Fallot and abnormal pulmonary venous return, which is suggestive of a partial expression of a cardiac laterality defect, has never been described in patients with mutations in the Ras/MAPK pathway.
The risk of cancer is reported to be rare in CFCS patients.2 None of our patients showed malignancy, and only a single patient with MEK1 mutation (Y130C) has been reported with a hepatoblastoma.25 Consistent with this finding, no somatic mutation in MEK1 gene has been observed in hepatoblastoma or in any other solid tumors (http://www.sanger.ac.uk/genetics/CGP/cosmic/). Similarly, the somatically acquired V600E amino acid substitution of BRAF occurs at high frequency in cancer, but it has not been observed among patients with CFCS. Accordingly, only two CFCS patients with BRAF mutations have been reported with malignancy.4, 26, 27 These data would indicate that CFCS syndrome should not be regarded as a cancer-predisposing condition.
In conclusion, our data confirm that mutations in MEK1 gene are more frequent than those in MEK2. CFCS patients heterozygous for MEK1 or MEK2 mutation genes display a phenotype well overlapping that observed in the general CFCS population and in patients with BRAF mutations, including dysmorphic facial features, mental retardation, development delay and ectodermal abnormalities. However, the frequency of CHDs, particularly mitral valve and septal defects, and of ocular anomalies seems lower among MEK1/MEK2 mutation-positive patients compared with patients carrying a BRAF gene mutation.
References
Roberts A, Allanson J, Jadico SK et al: The cardiofaciocutaneous syndrome. J Med Genet 2006; 43: 833–842.
Niihori T, Aoki Y, Narumi Y et al: Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet 2006; 38: 294–296.
Narumi Y, Aoki Y, Niihori T et al: Molecular and clinical characterization of cardio-facio-cutaneous (CFCS) syndrome: overlapping clinical manifestations with Costello syndrome. Am J Med Genet A 2007; 143: 799–807.
Rodriguez-Viciana P, Tetsu O, Tidyman WE et al: Germiline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 2006; 311: 287–290.
Schubbert S, Zenker M, Rowe SL et al: Germline KRAS mutations cause Noonan syndrome. Nat Genet 2006; 38: 331–336.
Schubbert S, Shannon K, Bollag G : Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 2007; 7: 295–308.
Tartaglia M, Mehler EL, Goldberg R et al: Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 2001; 29: 465–468.
Tartaglia M, Pennacchio LA, Zhao C et al: Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet 2007; 39: 75–79.
Carta C, Pantaleoni F, Bocchinfuso G et al: Germline missense mutations affecting KRAS isoform B are associated with a severe Noonan syndrome phenotype. Am J Med Genet 2006; 79: 129–135.
Pandit B, Sarkozy A, Pennacchio LA et al: Gain of function RAF1 mutations cause Noonan and Leopard syndromes with hypertrophic cardiomyopathy. Nat Genet 2007; 39: 1007–1012.
Razzaque MA, Nishizawa T, Komoike Y et al: Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet 2007; 39: 1013–1017.
Roberts A, Araki T, Swanson KD et al: Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet 2007; 1: 70–74.
Digilio MC, Conti E, Sarkozy A et al: Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet 2002; 71: 389–394.
Aoki Y, Niihori T, Kawame H et al: Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet 2005; 37: 1038–1040.
Bentires-Alj M, Kontaridis MI, Neel BG : Stops along the RAS pathway in human genetic disease. Nat Med 2006; 12: 283–285.
Nava C, Hanna N, Michot C et al: Cardio-facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway: genotype-phenotype relationships and overlap with Costello syndrome. J Med Genet 2007; 44: 763–771.
Schulz AL, Albrecht B, Arici C et al: Mutation and phenotypic spectrum in patients with cardio-facio-cutaneous and Costello syndrome. Clin Genet 2008; 73: 62–70.
Nyström AM, Ekvall S, Berglund E et al: Noonan and cardio-facio-cutaneous syndromes: two clinically and genetically overlapping disorders. J Med Genet 2008; 45: 500–506.
Shaul YD, Seger R : The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta 2007; 1773: 1213–1226.
Yoon G, Rosenberg J, Blaser S, Rauen KA : Neurological complications of cardio-facio-cutaneous syndrome. Dev Med Child Neurol 2007; 49: 894–899.
Gripp KW, Lin AE, Nicholson L et al: Further delineation of the phenotype resulting from BRAF or MEK1 germline mutations helps differentiate cardio-facio-cutaneous syndrome from Costello syndrome. Am J Med Genet A 2007; 143: 1472–1480.
Armour CM, Allanson JE : Further delineation of cardio-facio-cutaneous syndrome: clinical features of 38 individuals with proven mutations. J Med Genet 2008; 45: 249–254.
Kavamura MI, Peres CA, Alchorne MM, Brunoni D : CFC index for the diagnosis of cardiofaciocutaneous syndrome. Am J Med Genet 2002; 112: 12–16.
Marino B, Digilio MC, Toscano A, Giannotti A, Dallapiccola B : Congenital heart diseases in children with Noonan syndrome: an expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr 1999; 135: 703–706.
Al-Rahawan MM, Chute DJ, Sol-Church K et al: Hepatoblastoma and heart transplantation in a patient with cardio-facio-cutaneous syndrome. Am J Med Genet A 2007; 143: 1481–1488.
Van Den Berg H, Hennekam RC : Acute lymphoblastic leukaemia in a patient with cardiofaciocutaneous syndrome. J Med Genet 1999; 36: 799–800.
Makita Y, Narumi Y, Yoshida M et al: Leukemia in cardio-facio-cutaneous (CFC) syndrome: a patient with a germline mutation in BRAF proto-oncogene. J Pediatr Hematol Oncol 2007; 29: 287–290.
Acknowledgements
We thank the patients and families who participated in this study and the physicians who referred the individuals. This study was supported by grants from the Italian Ministry of Health, Ricerca Corrente 2008 (BD and FL), Telethon-Italy grant GGP07115 (MT), FIRB 2006 (BD), and ‘Programma di Collaborazione Italia-USA/malattie rare’ (AS and MT). VC was supported by a fellowship from Associazione ONLUS ‘Morgan Di Gianvittorio per la cura e la ricerca nei tumori e leucemie in età pediatrica’.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dentici, M., Sarkozy, A., Pantaleoni, F. et al. Spectrum of MEK1 and MEK2 gene mutations in cardio-facio-cutaneous syndrome and genotype–phenotype correlations. Eur J Hum Genet 17, 733–740 (2009). https://doi.org/10.1038/ejhg.2008.256
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.256
Keywords
This article is cited by
-
Intrinsically active MEK variants are differentially regulated by proteinases and phosphatases
Scientific Reports (2018)
-
Atrial ERK1/2 activation in the embryo leads to incomplete Septal closure: a novel mouse model of atrial Septal defect
Journal of Biomedical Science (2017)
-
Diagnosis of Noonan syndrome and related disorders using target next generation sequencing
BMC Medical Genetics (2014)
