
Abstract
Xq28 duplications encompassing MECP2 have been described in male patients with a severe neurodevelopmental disorder associated with hypotonia and spasticity, severe learning disability and recurrent pneumonia. We identified an Xq28 duplication in three families where several male patients had presented with intestinal pseudo-obstruction or bladder distension. The affected boys had similar dysmorphic facial appearances. Subsequently, we ascertained seven further families where the proband presented with similar features. We demonstrated duplications of the Xq28 region in five of these additional families. In addition to MECP2, these duplications encompassed several other genes already known to be associated with diseases including SLC6A8, L1CAM and Filamin A (FLNA). The two remaining families were shown to have intragenic duplications of FLNA only. We discuss which elements of the Xq28 duplication phenotype may be associated with the various genes in the duplication. We propose that duplication of FLNA may contribute to the bowel and bladder phenotype seen in these seven families.
Similar content being viewed by others


Introduction
In 2005, Meins et al1 described a submicroscopic duplication of Xq28 in a male patient with Rett syndrome-like features including developmental regression and stereotypic hand movements. The authors also noted dysmorphic features with a narrow mid-face. Subsequently, further patients with duplications of the same region have been described.2, 3, 4, 5, 6, 7 Van Esch et al2 identified an Xq28 duplication in a male patient from a large X-linked mental retardation family using array comparative genomic hybridisation (aCGH). This prompted them to screen 17 further male patients with similar phenotypes and led to the identification of similar duplications in male patients from three families. The consistent features of the phenotype described were severe mental retardation, axial hypotonia, seizures, acquired spasticity and frequent infections. It was noted that some of the affected male patients had hypotonic facies, a flat nasal bridge and prominent ears. Over 50 patients have now been reported with Xq28 duplications varying in size from 0.2 Mb to larger scale duplications extending beyond Xq27.8, 9, 10 Most have included the MECP2 and L1CAM genes. It has been suggested that the Xq28 duplication phenotype might be solely due to duplication of the MECP2 gene as patients with duplications involving this region alone have been reported with the same classical phenotype.1, 4, 5 We initially identified duplications in Xq28 using the multiplex ligation-dependent probe amplification (MLPA) technique in male patients from three families who presented with X-linked mental retardation. We noted that functional intestinal or bladder obstruction and characteristic ‘pinched’ facies were prominent features of affected male patients. We subsequently carried out more detailed analysis of the Xq28 region using further extended MLPA test kits or CGH using either 250K SNP chips or an Agilent array in the original three families and in seven further families with similar bowel and or facial phenotypes. Gargiulo et al11 reported Filamin A (FLNA) mutations in X-linked families with intestinal pseudo-obstruction; four of the families in our study (families 1, 5, 8 and 9) had previously been investigated by this group for FLNA mutations with negative results. We confirmed that affected male patients from these four families had a duplication of the FLNA gene and hypothesise that FLNA duplications may be associated with the presence of bowel dysfunction in patients with Xq28 duplications. Bladder dysfunction and/or dilatation was also observed in 4 of our 20 patients and although it is clearly associated with Xq 28 duplication, it seems to be a more variable feature of the phenotype and may not be specifically associated with FLNA involvement.
Materials and methods
Case reports
The main clinical features are also summarised in Table 1.
Family 1
Sibling 1. The second child born to a healthy couple who already had a healthy daughter. He was born at full term with a birth weight of 3 kg (>9th centile). He was hypotonic and failed to thrive in the neonatal period. He had severe constipation, required regular enemas and had a distended abdomen. In the first year, he developed seizures and had frequent chest infections. He never learned to sit or roll. He died at the age of 2½ years of bronchopneumonia and right-sided cardiac failure. At autopsy, generalised distention of the large bowel, bladder and ureters were noted. There was no major brain abnormality. Photographs show that he had a small narrow nose and a prominent forehead.
Sibling 2 (proband). The full brother of case 1, born at full term with a birth weight of 2.6 kg (2nd centile). In the neonatal period, he had recurrent intermittent bowel obstruction and was profoundly hypotonic. Rectal biopsy demonstrated normal neurons in the submucous and myenteric plexuses. Occasional neurons were noted to show nuclear and cytoplasmic vacuolation. He required daily enemas, had marked abdominal distention (Figure 1) and frequent chest infections. He had no speech and was profoundly delayed; he sat independently at 3 years of age. He had a small narrow nose and a prominent forehead with visible veins. His weight and length were below the 3rd centile but the head circumference was between the 5th and 10th centiles. He died of pneumonia at 9 years of age. At autopsy, there was marked distension of the intestines. The brain weight was within the normal range but histological examination showed an increased number of neurons that were larger than normal in the granular layer of the cerebral cortex, basal ganglia, hippocampus and lateral geniculate nuclei. The cerebellum showed marked enlargement of the dentate nucleus.

Photograph of the proband from family 1 demonstrating extreme abdominal distension due to chronic intestinal pseudo-obstruction. This child also shows characteristic facial features with pinched nose and a prominent forehead.
Sibling 3. This boy had a different father to that of his two older brothers. He was born at full term with a birth weight of 2.6 kg (>2nd centile). Initially, he fed well and gained weight but was hypotonic. By 6 months, he was significantly developmentally delayed and failing to thrive. At 16 months, he was unable to sit without support and had no speech development. Constipation was severe requiring daily enemas. Facially, he had a pinched nose and prominent forehead with thin appearing skin. He required a gastrostomy and fundoplication because of frequent chest infections and reflux. He made hand-flapping and hand-wringing movements reminiscent of those seen in Rett syndrome. MECP2 mutation testing was negative. MRI scan of his head showed high signal intensity in the periventricular white matter adjacent to the posterior horns of the lateral ventricles. The ventricular size, configuration of the extracerebral CSF spaces, corpus callosum and cerebellum were normal.
Family 2
A 16-year-old girl attended clinic with her mother asking for ‘carrier testing’. Her two maternal half-brothers (siblings 1 and 2) and her maternal aunt's son all had severe/profound developmental delay, constipation, recurrent chest infections, allergic rhinitis and seizures. All three died around 20 years of age from pneumonia. No DNA samples were available. The mothers of the affected boys were shown to have markedly skewed X inactivation. The phenotypic similarities between this family and family 1 had been recognised by the clinicians concerned several years before the molecular diagnosis.
Sibling 1. He was born at term weighing approximately 2.7 kg. He spent 10 days in the special-care baby unit with poor feeding. He suffered from severe constipation. He had severe/profound mental retardation and had seizures from age 4 years. At 4 and 10 years, he functioned at a 10-month level with an intelligence quota (IQ) estimated to be 20. He never developed speech nor walked but was able to stand with support. He had subluxation of his hips. He had frequent hospital admissions for pneumonia. He remained small with height, weight and head circumference below 0.4th centiles. He was dysmorphic with a long thin face, down slanting palpebral fissures, a prominent nasal bridge and long narrow nose. His fingers and feet were long. He was described as double jointed at the shoulders and elbows. He was diagnosed with cerebral palsy because of increased tone. Dystonic movements were noted in his hands.
Sibling 2. He was born at term weighing 2.7 kg. Developmental delay was more severe than GD1. He smiled at 3 months, never sat unsupported and had no speech. He also suffered from extreme constipation, seizures, recurrent pneumonia and increased tone in his legs. He had four long bone fractures after minimal injury. His growth parameters and dysmorphic features were similar to his brother's.
Cousin 1. He was born after a complicated breech delivery. His development and growth was less delayed compared with his cousins'. He smiled at 4 weeks, sat at 7 months, crawled at 1 year and walked with a frame from 3 years. He never walked independently. His IQ was 30 at 13 years; however, at 18 years, he was said to be functioning at the level of a 6-year-old child. He had no speech, seizures from 12 years, extreme constipation and recurrent pneumonia. He had two long bone fractures. His growth parameters were within the normal range. Photographs demonstrated possible dystonic movements; his nose was narrow but he was not strikingly dysmorphic.
Family 3
Sibling 1 (proband). A male child born at 42 weeks gestation with a birth weight of 2.83 kg (9th centile) presented with neonatal hypotonia. He fed poorly and had oesophageal reflux for which he subsequently underwent a fundoplication and gastrostomy. He required catheterisation for urinary retention. He had several episodes of right heart failure, treated with digoxin and diuretics, and had repeated admissions for pneumonia during infancy. He made very little developmental progress, and at 6 months he remained hypotonic and was unable to sit unsupported. At the age of 16 months, all of his growth parameters were below the 3rd centile. He underwent extensive investigations including a full metabolic screen but no specific cause was identified for his problems. Serum lactate was slightly elevated at 3.19 mmol/l. An MRI scan showed delayed myelination and absence of the anterior part of the corpus callosum. An IVP revealed a large thick-walled bladder. He died from recurrent respiratory problems at 3 years of age.
Sibling 2. The second affected male child in this family was born at term weighing 2.9 kg (>9th centile). He was hypotonic at birth with a distended abdomen due to a large trabeculated bladder that emptied following catheterisation. He was dysmorphic with a narrow nose, deep-set eyes, small chin and unusual ears. He held his thumbs clasped across his palms. CT scan revealed partial agenesis of the corpus callosum and mild dilatation of the lateral, third and fourth ventricles. He required fundoplication for gastro-oesophageal reflux. He also required treatment for constipation. He had recurrent bouts of pneumonia and suffered from unexplained anaemia, prompting bone marrow examination that showed some red cell hypoplasia. He made very little developmental progress before his death from respiratory problems and cardiac failure at 10 months of age.
The mother of these two boys was healthy. She had three further healthy children. She had several male siblings who had died as infants. One of them was diagnosed with prune belly syndrome and another had a grossly distended bladder after birth.
Family 4
Proband. He was born by caesarean section for breech presentation at 38 weeks gestation, weighing 2700 g (2nd–9th centile). He was breastfed poorly. From 6 months, he needed a nasopharyngeal airway and had frequent chest infections and episodes of pneumonia. At 8 years, he requires oxygen because of low saturations. He was hyotonic, sat at 13 months, has never walked and has no words. He has severe constipation requiring daily suppositories. Seizures began at 6 years; EEG showed an excess of slow wave activity but no specific features and MRI scan showed cerebral hypoplasia/atrophy with increased size of the ventricles and incomplete myelination, and there was an abnormal periventricular white matter signal. His head circumference was on the 9th centile and his weight on the 50th centile. He had spasticity of his lower limbs with contractures at his hips and knees. He had deep-set eyes, a narrow nose and a prominent chin; his skin appeared pale and thin. He had cold red feet with poor circulation. He walked with support with a wide-based gait, and frequent hand flapping was observed. He was investigated extensively for a metabolic disorder with negative results. A diagnosis of Angelman syndrome had also been suggested, but methylation analysis of chromosome 15q11–13 and UBE3A mutation analysis were normal.
Family 5
Sibling 1. A male child born at full term to a healthy mother who had learning problems at school. There were normal birth parameters (OFC 33 cm, 9th centile; weight 3.160 kg, 25th centile; length 50 cm, 25th centile). He was noted to have a patent common mesentery. He had major feeding difficulties, chronic constipation and gastro-oesophageal reflux. He was significantly hypotonic but sat unaided at 9 months and walked at 24 months. He had postnatal growth retardation (−3 SD for weight and height). OFC was −0.5 SD. At 4 years, he could say a few words and was hyperactive. No spasticity was noted. His facial appearance and other clinical features were similar to those of the proband in this family (sibling 2, below). Cerebral MRI studies revealed partial agenesis of the posterior corpus callosum and cortical atrophy.
Sibling 2. He was born at term with normal birth parameters (OFC 33.5 cm, weight 2.800 kg and length 48 cm, all equal to or >9th centile). He had chronic constipation and gastro-oesophageal reflux. He was hypotonic, developed head control at 4 months and walked with aids at 30 months. He was diagnosed as having pseudointestinal obstruction following rectal biopsy, which ruled out Hirschsprung disease. He had frequent chest infections and exhibited dysautonomia in these episodes. He also had myoclonic epilepsy. At 3 years, his height and OFC were −2 SD. He was considered to have an autistic spectrum disorder, but he had no sleep disorder and no spasticity. He was dysmorphic with hypertelorism, frontal bossing, low nasal bridge, anteverted nares, everted lower lip and small wide-spaced teeth. Investigations including brain MRI and auditory-evoked potentials, chromosome studies and metabolic screening in plasma and CSF were all normal. Screening for carbohydrate-deficient glycoprotein disorders and RSK2 screening were also negative.
Half-sibling 3. The half-brother of siblings 1 and 2 with the same mother. He was born at term after a normal pregnancy. His birth parameters were normal with an OFC of 34 cm, weight 3700 g and length 51 cm (all between 25th and 50th centiles). He showed normal postnatal growth, but was hypotonic and did not gain head control until 4 months. He also has severe constipation.
Family 6
Proband. A male, the only child of healthy unrelated parents, was born at 37 weeks gestation by caesarean section with a birth weight of 1.800 g (<3rd centile). Prenatal ultrasound had shown urinary bladder dilatation; at birth he was noted to have right cryptorchidism and abdominal ultrasound confirmed megacystis with mild dilatation of the renal pelvis. He had severe feeding difficulties, growth retardation and hypotonia. For the first 2 years of life, he suffered from severe constipation. Barium radiographs of the upper and lower gastrointestinal tracts revealed dilated and hypotonic loops of the proximal portion of the large bowel and a reduction in the diameter of the distal portion. By the age of 5 years, bowel function had improved. He acquired head control at 4 months of age and was able to walk independently at 24 months of age. At 7 years, he has normal height and weight and a relatively small head circumference (−1 SD); he is severely retarded, has epilepsy, recurrent respiratory infections and gastro-oesophageal reflux. Speech is limited to two to three words and his behaviour is hyperactive. Cerebral MRI, at 2 years of age, showed enlargement of the cisterna magna, mild hypoplasia of the cerebellar vermis, mild hypoplasia of the corpus callosum and delayed myelination. Echocardiogram showed a bicuspid aortic valve with mild valvular insufficiency. His face is hypotonic, his forehead is prominent and he has a narrow nose and small mouth with flat malar regions. His ears appear large.
Family 7
Proband. A male, the only child born to healthy unrelated parents at 37 weeks by caesarian section performed for foetal bradycardia. Birth measurements were around the 3rd centiles for length, weight and head circumference. Prenatal ultrasound revealed intrauterine growth retardation from the twentieth week of pregnancy. In the first 2 years of life, he suffered from gastro-oesophageal reflux. He achieved head control at the age of 12 months and independent sitting at the age of 2 years. At 6 years, his height, weight and head circumference are all below the 3rd centiles and he is severely mentally retarded. He is hypotonic and not able to stand or walk independently and has no speech. He has had recurrent respiratory infections and has severe chronic constipation. Despite a high-fibre diet and the use of glycerin suppositories, he has his bowels open only once or twice a week. He has plagiocephaly, a prominent forehead, sparse eyebrows and short palpebral fissures. His malar regions appear flat and his nose small; the philtrum is short and he has a pointed chin. MRI revealed periventricular leucomalacia. Ophthalmological examination showed bilateral astigmatism and exotropia.
Family 8
A family with three affected male patients with chronic neurogenic intestinal pseudo-obstruction, thrombcytopaenia, PDA, malrotation and characteristic facies and without mental retardation previously reported.12 Linkage was demonstrated to markers at Xq28.
Family 9
Proband. The first child born to a 35-year-old mother and a 32-year-old father. In pregnancy, dilated loops of bowel and a large cisterna magna were detected by ultrasound scan. He was born at 40 weeks gestation with a birth weight of 3.555 kg, length 51 cm and OFC 36 cm (all ∼50th centile). A single umbilical artery and mild dilatation of the renal pelvis were present. He had a wide patent ductus arteriosus and a patent foramen ovale leading to heart failure at 13 days. He underwent ligation of the patent ductus at 16 days. He developed intestinal obstruction and was found to have bowel malrotation with Ladd's bands. These were divided, but his intestinal obstruction persisted and a second operation revealed dilatation of the small bowel without mechanical cause. He was treated by ileostomy and parenteral nutrition. Intestinal biopsies excluded Hirschsprung's disease but suggested a neurogenic cause for the bowel obstruction. Thrombocytopaenia was also present (43–97 × 109/l) with the presence of giant platelets and anisomegakaryocytes. He was not facially dysmorphic. His features were considered very similar to those of family 6 before the current investigations were undertaken.
Family 10
Proband. The second child born to healthy parents, who is currently aged 20 years. He was born following a normal pregnancy, but development was delayed and he walked at the age of 6 years. He has never developed speech. He had feeding difficulties and in the first years of life he had chronic diarrhoea. He has poor coordination, sleep disorder and autistic features. He has a seizure disorder that began at the age of 13 years. He has hypothyroidism and has suffered from frequent severe respiratory infections. More recently, he has had two episodes of septicaemia, one resulting in amputation of his left leg. On clinical examination at the age of 20 years he has facial hypotonia, down-slanting palpebral fissures, hypoplastic alae nasi, a high-arched palate, arachnodactyly, joint hyperlaxity and scoliosis.
Sibling 1. He is the brother of the proband. He showed severe psychomotor delay from birth and was noted to have dysmorphic facial features. He suffered from severe constipation, and despite a high-fibre diet and enemas, he defaecated only twice a week. He died at the age of 18 months following a severe infection.
The proband had three other siblings. Two of these were born healthy (one subsequently died as a result of trauma). The remaining sibling, a girl, who was the twin of sibling 1 above, had a mild learning difficulty and attention problems.
Cytogenetic and molecular analyses
MLPA was performed on genomic DNA isolated from peripheral blood samples using the Gentra Puregene kit (Qiagen, Valencia, CA, USA). The MLPA method used was as described by Schouten et al,13 using MLPA kits purchased from MRC-Holland (Amsterdam, the Netherlands). Initial MLPA studies of the Xq28 region were performed using the P015C kit and included probes for four exons of MECP2, IRAK1, L1CAM, IDH3G and SLC6A8, alongside 12 X chromosome control probes and four autosomal control probes. Subsequently, MLPA of the FLNA gene was performed using the P049 kit and included probes for FMR2, IDS, three exons of PNCK, 10 exons of SLC6A8, three exons of BCAP31, two exons of ABCD1, IDH3G, four exons of L1CAM, IRAK1, two exons and one intron of MECP2, three exons of FLNA, two exons of GDI1, two exons of FVIII and SYBL1, alongside six chromosome X control probes.
MLPA was performed as follows: 200 ng of DNA was denatured and hybridised with the probe mix at 60°C overnight. The following morning, the paired probes were ligated using ligase-65 at 54°C for 15 min. This was followed by a PCR using a common M13 primer pair and the following PCR conditions; 95°C for 30 s, 60°C for 30 s and 72°C for 1 min for 35 cycles. The products were run on an ABI 3130 Genetic Analyser and analysed using the Genemapper 4.0 software (Applied Biosystems, Foster City, CA, USA). Data were normalised and analysed by peak heights, using the analysis spreadsheets from the National Genetics Reference Laboratory (NGRL, Manchester, UK).
An affected male patient from each of the families 1, 3, 4, 5, 8 and 9 was tested for copy number variations at the Xq28 region. In family 2, where no DNA was available from an affected male patient, the mother of cases 1 and 2 was tested. Where possible, maternal samples were also tested. Results were confirmed using an Affymetrix SNP analyser with a 250 kb resolution. Patients from families 6, 7 and 10 were studied by aCGH using an Agilent 44B array at a resolution of approximately 100 kb.
Results
Affected male patients in all families had previously been shown to have normal routine karyotypes. In family 7, FISH with subtelomeric probes revealed the absence of signal of the probe DXYS129 (distal PAR1) at Yp and the presence of three signals of the probe ESTCdYC07, with two signals at Xq and Yq and a third signal at Yp. The imbalance was at first interpreted as recombinant chromosome [ish der(Y)inv(Y)(p11;q22)(DXYS129−ESTCdyC07++)].
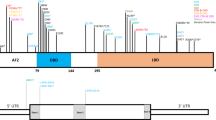
Investigation of affected male patients from families 1, 3, 4, 5, 8 and 9 and the mother of family 2 using the PO15C kit demonstrated duplications in families 1–5. Further studies using the P049 kit, which also included the FLNA gene, identified duplications in all of these families (Figure 2). Patients from families 6, 7 and 10 were shown to have Xq28 duplications using the Agilent 44B array. Detailed mapping of the extent of the deletions has not yet been undertaken. In families 8 and 9, the duplications involved only the FLNA gene where intragenic duplications of exons 4 and 11, but not exon 40, were demonstrated. Affected male patients from each of these two families had previously been screened for point mutations within the FLNA gene with negative results. All of these results were confirmed using the X chromosome SNP array. Investigation of the available mothers confirmed that the mothers in families 1, 2, 3, 5, 6 and 10 were obligate carriers. In family 10, the maternal grandmother and the mentally retarded sister were also carriers of the same rearrangement, whereas the unaffected sister did not show the same imbalance. In families 4 and 7, the duplications appeared to have arisen de novo as no duplications were demonstrated in the mother. X-inactivation testing on blood samples from the female patients in family 10 showed skewed inactivation in all the female patients carrying the Xq28 duplication, with the inactive X chromosome being the duplicated one. Further studies of family 7 failed to define the break points of the Yp deletion because of the lack of terminal Yp oligonucleotides on the platform used. This rearrangement most likely involves the distal PAR1 region encompassing the SHOX gene as the proband has a short stature. FISH analysis using a specific Xq28 probe (RP11-119A22) confirmed the translocation of the duplicated region to the short arm of chromosome Y.

Schematic representation of the Xq28 region to demonstrate the extent of the duplications in families 1–10. Families 1, 2, 3, 4, 5, 8 and 9 were analysed using MLPA with the PO15C and the PO49 kits. The regions covered by both of these are indicated at the top of the diagram in relation to the position of known genes within the Xq28 region. The MLPA results in the above families are shown, with filled blocks indicating the duplicated regions and open blocks indicating normal copy number. When repeated, the MLPA result in family 3 has suggested that some regions may be triplicated and others duplicated, and this is currently the subject of further investigation. Families 6, 7 and 10 were studied by array CGH using the Agilent 44B array at a resolution of approximately 100 Mb, and the extent of the duplications is shown in the lower part of the diagram. These varied in size, the largest being approximately 4 Mb (150.49–154.5), the intermediate 1.3 Mb (152–153.4) and the smallest 650 kb (152.5–153.13).
Discussion
Over 50 male patients with duplications of the Xq28 region have now been reported; several previous authors have reviewed the sizes of the duplications and commented on possible correlation between the phenotype and the genes included in the duplicated regions.4, 7 Although spasticity, severe learning disability, axial hypotonia and frequent chest infections have been common findings within this group of patients, gastrointestinal symptoms have been much rarer. Gastro-oesophageal reflux has been mentioned as a feature in some patients,3 but other bowel symptoms have been infrequent. In the report by Meins et al,1 one patient had suspected gluten enteropathy because of poor weight gain but no other bowel symptoms.
Many neurological and skeletal phenotypes have been associated with FLNA mutations.14 Missense mutations, generally thought to lead to gain of function, cause the skeletal phenotypes of otopalatodigital syndrome, Melnick–Needles syndrome and frontometaphyseal dysplasia. FLNA loss-of-function mutations are associated with the PVNH phenotype and are usually embryonically lethal in male patients.15 Virtually, all of these mutations are frameshift or truncating and likely to be subject to nonsense-mediated RNA decay. There have been two reports of surviving male patients with PVNH associated with duplications of the Xq28 region,15, 16 suggesting that the amount of FLNA is critical for neuronal migration, and either increased or decreased expression can interfere with the normal migration process. This hypothesis is also supported by animal studies.17
Chronic intestinal pseudo-obstruction has not been a prominent feature in patients with FLNA mutations who were ascertained because they had PVNH, and this may be because they often do not survive. Interestingly, one heterozygous female patient was noted to have problems with gastrointestinal motility.15 Surviving male patients with milder FLNA mutations, where some FLNA function is retained, have been described18 but again, gastrointestinal problems were not noted as a prominent feature.
Gargiulo11 studied a family that was ascertained because of intestinal pseudo-obstruction, and identified a 2-bp deletion in FLNA. Affected members of this family do not appear to have been screened for the presence of PVNH. Although the deletion caused a frameshift, the presence of an alternative ATG site 22 codons downstream of the initial ATG enabled the production of a shorter FLNA protein. The authors thus hypothesised that the first 27 FLNA residues might be crucial for enteric neuron structure and function. Alternatively, the bowel symptoms could have resulted from an overall reduced FLN expression. Hehr19 described a further patient with PVNH and severe constipation who had an FLNA splice-site mutation leading to the production of both a normal and a shorter mRNA. These findings might be consistent with either of the above hypotheses. FLNA protein expression in our families has not yet been studied in detail, and thus suggestions as to how the intestinal phenotype is caused are purely speculative; one hypothesis is that increased FLNA dosage significantly compromises enteric neuron development in addition to affecting neuronal migration. The findings in Hehr's patient suggest that the bowel problems may, like neuronal migration, be affected by both under- and overexpression of FLNA. Phenotyping of the reported patients with FLNA mutations or duplications has not always been comprehensive. Patients ascertained through chronic intestinal pseudo-obstruction have not always been investigated for PVNH and vice versa. Studies of filamin expression, more detailed phenotyping and careful scrutiny of the MRI images of patients with both FLNA mutations and duplications should help to resolve this issue.
OMIM entry 300048 concerns the phenotype of X-linked chronic idiopathic intestinal pseudo-obstruction (CIIP) associated with additional features including patent ductus arteriosus, short bowel, malrotation and coagulopathy. This entity includes the family reported by Fitzpatrick et al12 (our family 8), who had all of the above features, along with a similar patient reported by Pollock et al.20 Two further cases were reported by Harris et al,21 and the Italian family with CIIP originally reported by Aurecchio et al,22 and then investigated by Gargiulo,11 is included. Although this Italian kindred was subsequently identified to have a loss-of-function mutation in FLNA, no mutation was identified in Fitzpatrick's family or in our family 9. It is not clear whether FLNA was studied in the patient reported by Pollock et al.20 The presence of thrombocytopaenia with large platelets and the absence of a filamin mutation set these two families apart from Gargiulo's kindred. Their duplication detected with the P049 MLPA kit demonstrated only duplication of exons 4 and 11 of the FLNA gene, and they were the only patients of our cohort in which thrombocytopaenia was a feature. Neither of the patients from families 8 and 9 had mental retardation, hypotonia or spasticity, and they were not facially so dysmorphic as the patients with large duplications. Thrombocytopaenia and cardiac defects are clearly not confined to patients with intragenic FLNA duplications; however, as Parrini et al23 reported that insufficiency of the aortic valve, PDA or idiopathic thrombocytopaenia were present in 20 of the 35 patients with FLNA mutations, they ascertained in their study of PVNH. Vascular events such as cerebrovascular accidents were also documented in female patients heterozygous for an FLNA mutation and a hemizygous male patient had a neonatal haemorrhage and maturation arrest of the bone marrow.15 There is clear demonstration, therefore, of the association between PVNH, CIIP, cardiac defects and thrombocytopaenia in patients with FLNA point mutations and intragenic duplications. A systematic search for these haematological and vascular features in the group of patients with larger Xq duplications may help to delineate the thrombocytopaenia and bone marrow phenotype further.
Unger et al24 reported a patient with a missense mutation in FLNA and a phenotype suggestive of FG syndrome. This child had presented with relative macrocephaly, an unusual head shape and severe and chronic constipation. He had a prominent forehead with a frontal cowlick, a small mouth and simple unfolded ears. Until recently, FG syndrome has been considered as a group of genetically heterogeneous X-linked conditions, and reported affected male patients have had diverse phenotypes, mainly including mental retardation, macrocephaly and severe constipation. Since the finding of MED12 mutations in the original FG family25 and Unger's report of an FLNA mutation, more distinctive phenotypes for FG syndrome have emerged. It is possible that some of the ‘FG’ patients without MED12 or FLNA mutations may have FLNA duplications. Interestingly, FG syndrome had been considered as a possible diagnosis in family 1.
Few authors have commented in great detail on the facial gestalt of the Xq28 duplication patients. Gargiulo for example, concentrated on the intestinal phenotype rather than including a description or photographs of facial features. We believe that there is a recognisable face (Figure 3). Families 1 and 2 were recognised as having the same syndrome on the basis of facial features as well as bowel symptoms several years before molecular diagnosis. Patient 4 was originally referred with a possible diagnosis of Angelman syndrome, but on the basis of his facial appearance, MECP2 duplication testing was undertaken. Most of our patients had a narrow ‘pinched’ appearance of the nose with a translucent quality of the skin and prominent veins. The eyes appeared deep-set and the chin prominent. The lower lip was frequently everted and small; widely spaced teeth were noted in some patients. A frontal cowlick was often present. We would agree with Van Esch et al2 that the ears appear prominent and the face hypotonic, though a flat nasal bridge tended to be present only in younger patients. Although microcephaly has frequently been reported with Xq28 duplications, this is not universally the case and several of our patients had head circumferences within the normal range even though their other growth parameters were impaired. This relative macrocephaly was associated with a prominent appearance of the forehead. An asymmetric skull shape has been commented upon in several patients both within our cohort and in the literature. Our patients looked remarkably similar to patient 3 of Smyk et al4 and patient II2 reported by Friez5. The Xq28 facial gestalt seems to be more recognisable from mid-childhood onwards and less recognisable in patients who have associated microcephaly. MRI brain scans had been carried out on several of our patients as detailed above. None of the patients were reported to have the classical appearance of PVNH, although abnormal periventricular white matter was reported in some (see Table 1), and these scans deserve further scrutiny. Abnormalities of the corpus callosum and delayed myelination were frequently reported and these features were also mentioned in the male patient with a duplication reported by Fox.15

Composite of faces showing, from left to right, two siblings from family 1, one from family 2 and the proband from family 4. All demonstrate similar hypotonic facies, narrow nose, prominent forehead and deep-set eyes. The mouth and chin are relatively small and the skin is thin with visible veins.
In summary, we propose that duplication of the Xq28 region is a significant cause of intestinal pseudo-obstruction, especially in male patients presenting with severe learning difficulties and hypotonia, and that this and the presence of characteristic facial features are helpful in recognition of the phenotype. Though Xq28 duplications will, for the most part, contain several genes, our observations suggest that it is the involvement of the FLNA gene that appears to be responsible for the presence of intestinal pseudo-obstruction. This is corroborated by evidence from previously reported patients with FLNA mutations and by animal studies. The occurrence of additional features including patent ductus arteriosus, short bowel, malrotation, thrombocytopaenia, maturation arrest of the bone marrow and agenesis of the corpus callosum, in addition to PVNH and CIIP, suggests that duplication of FLNA is acting as a hypomorphic mutation with pleiotrophic effects in affected male patients. Male patients with larger duplications had the additional neurological features of mental retardation, hypotonia or spasticity, but it seems feasible that many of the phenotypic features of the Xq28 duplication may be due to FLNA involvement alone.
References
Meins M, Lehmann J, Gerresheim F et al: Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet 2005; 42: e12.
Van Esch H, Bauters M, Ignatius J et al: Duplication of the MECP2 Region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet 2005; 77: 442–453.
Sanlaville D, Prieur M, de Blois M-C et al: Functional disomy of the Xq28 chromosome region. Eur J Hum Genet 2005; 13: 579–585.
Smyk M, Obersztyn E, Nowakowska B et al: Different-sized duplications of Xq28, including MECP2, in three males with mental retardation, absent or delayed speech, and recurrent infections. Am J Med Genet B Neuropsychiatr Genet 2008; 147B: 799–806.
Friez MJ, Jones JR, Clarkson K et al: Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics 2006; 118: e1687–e1695.
Lugtenberg D, de Brouwer APM, Kleefstra T et al: Chromosomal copy number changes in patients with non-syndromic X linked mental retardation detected by array CGH. J Med Genet 2006; 43: 362–370.
del Gaudio D, Fang P, Scaglia F et al: Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med 2006; 8: 784–792.
Lachlan KL, Collinson MN, Sandford ROC, van Zyl B, Jacobs PA, Thomas NS : Functional disomy resulting from duplications of distal Xq in four unrelated patients. Hum Genet 2004; 115: 399–408.
Lammer EJ, Punglia DR, Fuchs AE, Rowe AG, Cotter PD : Inherited duplication of Xq27.2 → qter: phenocopy of infantile Prader–Willi syndrome. Clin Dysmorphol 2001; 10: 141–144.
Novelli A, Bernardini L, Salpietro DC et al: Disomy of distal Xq in males. Am J Med Genet A 2004; 128A: 165–169.
Gargiulo A, Auricchio R, Barone MV et al: Filamin A is mutated in X-linked chronic idiopathic intestinal pseudo-obstruction with central nervous system involvement. Am J Hum Genet 2007; 80: 751–758.
FitzPatrick DR, Strain L, Thomas AE et al: Neurogenic chronic idiopathic intestinal pseudo-obstruction, patent ductus arteriosus, and thrombocytopenia segregating as an X linked recessive disorder. J Med Genet 1997; 34: 666–669.
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G : Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002; 30: e57.
Robertson SP : Filamin A: phenotypic diversity. Curr Opin Genet Dev 2005; 15: 301–307.
Fox JW, Lamperti ED, Eksioğlu YZ et al: Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron 1998; 21: 1315–1325.
Fink JM, Dobyns WB, Guerrini R, Hirsch BA : Identification of a duplication of Xq28 associated with bilateral periventricular nodular heterotopia. Am J Hum Genet 1997; 61: 379–387.
Nagano T, Morikubo S, Sato M : Filamin A and FILIP (Filamin A-Interacting Protein) regulate cell polarity and motility in neocortical subventricular and intermediate zones during radial migration. J Neurosci 2004; 24: 9648–9657.
Sheen VL, Dixon PH, Fox JW et al: Mutations in the X-linked Filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum Mol Genet 2001; 10: 1775–1783.
Hehr U, Hehr A, Uvanik G, Phelan E, Winkler J, Reardon W : A filamin A splice mutation resulting in a syndrome of facial dysmorphism, periventricular nodular heterotopia, and severe constipation reminiscent of cerebro-fronto-facial syndrome. J Med Genet 2006; 43: 541–544.
Pollock I, Holmes SJK, Patton MA, Hamilton PA, Stacey TE : Congenital intestinal pseudo-obstruction associated with a giant platelet disorder. J Med Genet 1991; 28: 495–496.
Harris DJ, Ashcraft KW, Beatty EC, Holder TM, Leonidas JC : Natal teeth, patent ductus arteriosus and intestinal pseudo-obstruction: a lethal syndrome in the newborn. Clin Genet 1976; 9: 479–482.
Auricchio A, Brancolini V, Casari G et al: The locus for a novel syndromic form of neuronal intestinal pseudoobstruction maps to Xq28. Am J Hum Genet 1996; 58: 743–748.
Parrini E, Ramazzotti A, Dobyns WB et al: Periventricular heterotopia: phenotypic heterogeneity and correlation with Filamin A mutations. Brain 2006; 129: 1892–1906.
Unger S, Mainberger A, Spitz C et al: Filamin A mutation is one cause of FG syndrome. Am J Med Genet A 2007; 143A: 1876–1879.
Risheg H, Graham Jr JM, Clark RD et al: A recurrent mutation in MED12 leading to R961W causes Opitz–Kaveggia syndrome. Nat Genet 2007; 39: 451–453.
Acknowledgements
This study was supported in part by Central Manchester and Manchester Children's University Hospitals NHS Trust through funding from North West Development Agency and PRIN 2005, Fondazione Mariani and Fondazione CARIPLO. We are grateful to G Black and R Parveen, University of Manchester, L Gaunt, CMMC, SD Arrigo, Istituto Neurologico, C Besta, Milan, L Garavelli, Ospedale S Maria Nuova Reggio Emilia and S Bernasconi Università degli Studi, Parma, Italy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Clayton-Smith, J., Walters, S., Hobson, E. et al. Xq28 duplication presenting with intestinal and bladder dysfunction and a distinctive facial appearance. Eur J Hum Genet 17, 434–443 (2009). https://doi.org/10.1038/ejhg.2008.192
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.192
Keywords
This article is cited by
-
IRAK1 Duplication in MECP2 Duplication Syndrome Does Not Increase Canonical NF-κB–Induced Inflammation
Journal of Clinical Immunology (2023)
-
A brief history of MECP2 duplication syndrome: 20-years of clinical understanding
Orphanet Journal of Rare Diseases (2022)
-
Prune belly syndrome in surviving males can be caused by Hemizygous missense mutations in the X-linked Filamin A gene
BMC Medical Genetics (2020)
-
A sibship with duplication of Xq28 inherited from the mother; genomic characterization and clinical outcomes
BMC Medical Genetics (2017)
