
Abstract
The CRISPR/Cas9 system is an efficient gene-editing method, but the majority of gene-edited animals showed mosaicism, with editing occurring only in a portion of cells. Here we show that single gene or multiple genes can be completely knocked out in mouse and monkey embryos by zygotic injection of Cas9 mRNA and multiple adjacent single-guide RNAs (spaced 10-200 bp apart) that target only a single key exon of each gene. Phenotypic analysis of F0 mice following targeted deletion of eight genes on the Y chromosome individually demonstrated the robustness of this approach in generating knockout mice. Importantly, this approach delivers complete gene knockout at high efficiencies (100% on Arntl and 91% on Prrt2) in monkey embryos. Finally, we could generate a complete Prrt2 knockout monkey in a single step, demonstrating the usefulness of this approach in rapidly establishing gene-edited monkey models.
Similar content being viewed by others


Introduction
The CRISPR/Cas9 system has been used to generate genome modification in various species by directly injecting Cas9 mRNA and single-guide RNAs (sgRNAs) into pronuclear stage zygotes, leading to a DNA double-strand break (DSB) at a specified locus1,2. The DSB could then be repaired with error-prone non-homologous end joining, resulting in mutant animals carrying frame-shift mutations. However, the majority of gene-edited animals generated by this method showed gene-functional mosaicism, with gene disruption occurring in some cells but not others3,4. For phenotypic analysis of gene function, gene-edited animals with mosaicism required further serial crossbreeding to generate complete gene knockout animals. Such a procedure becomes rather laborious when deletion of multiple genes is required. The problem of mosaicism becomes even more serious when the CRISPR/Cas9 method is applied to large animals such as macaque monkeys, which have a long breeding cycle (5-6 years) and a small litter size (1 per birth)4,5.
Many previous studies have attempted to produce gene-modified animals without mosaicism in a single step, a procedure especially useful for large animals. These include injection of Cas9 mRNA and sgRNAs into the oocyte rather than the zygote6, injection of Cas9 protein7,8, or dual sgRNAs injection9,10. However, these approaches achieved complete biallelic knockout animals with rather low efficiency.
In this study, we show that single gene or multiple genes can be completely knocked out in all cells of nearly 100% of CRISPR/Cas9-injected mice and monkeys, using multiple sgRNAs. We demonstrate the usefulness of this approach in rapid screening of gene functions in F0 mice and in the rapid generation of monkeys with a complete knockout of a specific gene.
Results
Complete deletion of GFP in transgenic GFP embryos
We first tested whether using multiple rather than single sgRNAs could achieve complete gene knockout. To facilitate the estimation of mosaicism, we co-injected Cas9 mRNA and GFP-targeting sgRNAs into zygotes derived from wild-type (WT) female mice mated with homozygous Actin-EGFP transgenic males (with ubiquitous expression in all cells) and then analyzed GFP expression pattern at the blastocyst stage (Figure 1A). We designed four sgRNAs with target sites spaced 10-200 bps at the exon of the GFP gene, surmising that such short distances between adjacent sgRNAs could lead to a high frequency of large-fragment exon deletion rather than regular indels, resulting in more frequent complete gene knockout. We found that 31%-44% of the blastocysts derived from zygotes targeted with single-sgRNAs exhibited no GFP signal, and the rest showed mosaicism, with GFP signals in some blastomeres (Figure 1B and 1C). In control embryos without gene editing, all the blastocysts were GFP-positive (Figure 1B and 1C). By contrast, after injection of multiple (2, 3, or 4) sgRNAs that targeted GFP, we found that the GFP signal was eliminated in all injected blastocysts, indicating a GFP complete knockout (Figure 1B and 1C). Furthermore, multiple-sgRNA targeting showed no obvious deleterious effect on the embryo development which was comparable with embryos obtained by single-sgRNA targeting (Figure 1D).

Complete deletion of GFP in GFP-embryos by C-CRISPR. (A) Schematic diagram of sgRNA-targeting sites at GFP locus and experimental design. Cas9 mRNA and single or multiple sgRNAs-targeting GFP were injected into individual mouse zygotes and GFP signal was examined at blastocyst stage. (B) Images of GFP signal of blastocysts resulting from different forms of GFP targeting. Examples of GFP-negative blastocyst (green arrow, no GFP signal in any cells of the blastocyst) and GFP-positive blastocyst with mosaicism (red arrow, GFP signal in some cells of the blastocyst) are shown in the first image. The concentration of each sgRNA was shown in each group. Scale bar, 200 μm. (C) Histograms showing percentages of GFP-negative blastocysts (without mosaicism) resulting from different forms of GFP targeting. Two or more targeting sgRNAs (sgRNA-GFP-A+B, A+B+C, or A+B+C+D) resulted in higher percentages of GFP-negative blastocysts than single sgRNA targeting (sgRNA-GFP-A, B, C, or D). Number above the bar, total number of blastocysts counted (***P < 0.001, χ2 test). The concentration of each sgRNA was shown at the bottom. (D) Blastocyst rate of embryos resulting from different GFP targeting. Number, total number of zygotes injected. The concentration of each sgRNA was shown at the bottom.
We next asked whether the sgRNA concentration could affect the efficiency of complete GFP knockout. Whereas in the above experiments we had injected sgRNAs at 50 ng/μl for each sgRNA, we now injected 4 sgRNAs (sgRNA-GFP-A+B+C+D) at 12.5 ng/μl for each sgRNA, or injected a single sgRNA (sgRNA-GFP-B) at 10, 100, and 200 ng/μl, respectively (Figure 1B). We found that all embryos injected with the 4 sgRNAs at this lower concentration exhibited no GFP signal whereas embryos injected with 1 sgRNA at different concentrations showed GFP mosaicism (Figure 1B and 1C). These results indicate that GFP complete knockout is not due to the increase of the concentration of sgRNAs, but to multiple-sgRNA targeting.
Complete deletion of Tyr gene in mice
To further test whether an endogenous gene with two alleles could also be completely knocked out by this approach, we targeted the Tyrosinase gene (Tyr, for pigmentation) at the zygote stage, and produced gene-edited mice via implantation of two-cell embryos (Figure 2A). We found that targeting a single exon with 3 or 4 sgRNAs (sgRNA-Tyr-B+C+D or sgRNA-Tyr-B+C+D+E) resulted in 100% of albino mice (Figure 2A-2C), indicating complete Tyr knockout in all gene-edited mice. By contrast, 1 sgRNA or 2 sgRNAs targeting one exon (sgRNA-Tyr-D or sgRNA-Tyr-C+D), or 2 sgRNAs targeting two introns flanking one exon (sgRNA-Tyr-A+F) resulted in 26%-77% of gene-edited mice with mosaicism of pigmentation (Figure 2A-2C). Furthermore, multiple-sgRNA targeting showed no obvious deleterious effect on either embryo development or birth rate of gene-edited mice, compared with single-sgRNA targeting (Figure 2D). The gene-modified mice created by 4-sgRNA targeting (sgRNA-Tyr-B+C+D+E) showed normal reproductive ability when mated to ICR (albino) mice and all the progeny were albino, indicating complete knockout of Tyr in all germ cells of these gene-modified mice (Figure 2E).

One-step generation of Tyr complete knockout mice by C-CRISPR. (A) Schematic diagram of sgRNA-targeting sites at Tyr locus. The space between each sgRNA is from the end of last base of one sgRNA to the first base of another sgRNA (including PAM sequence). (B) Representative results of pigmentation phenotypes of mice resulting from Tyr targeting. Green arrowhead, albino; red arrowhead, mosaic pigmentation. (C) Histograms showing the percentage of albino mice resulting from Tyr targeting. Two or more targeting sgRNAs (sgRNA-Tyr-C+D, B+C+D, or B+C+D+E) resulted in higher percentage of albino mice than single-sgRNA targeting (sgRNA-Tyr-C or D). Number, total number of mice counted (***P < 0.001, χ2 test). (D) Birth rate of mice resulting from Tyr targeting. Number, total number of embryos transferred. (E) Reproductive ability and germline transmission of Tyr knockout mice by C-CRISPR. Five gene-modified mice (sgRNA-Tyr-B+C+D+E) were mated with WT ICR mice individually. Each pair of mice gave birth to two litters of pups. (F) Albino mice resulting from sgRNA-Tyr-B+C+D+E targeting were sequenced. DNA was isolated from tails of albino mice and PCR amplified. #2 mice contained large-fragment exon deletion and indels. LED, large-fragment exon deletion; Indels, insertion, or deletion of bases. Number above each column, total TA clones sequenced. (G and H) Percentages of different mutation types (G) and genotypes (H) of blastocysts resulting from Tyr targeting. Number, total TA clones sequenced and analyzed. WT, wild-type allele. Complete KO, blastocyst with complete knockout mutations; Incomplete KO, blastocyst with WT allele and knockout mutations. Number, total blastocysts counted. (I) Size distribution of deletions or insertions in blastocysts with Tyr targeting. (J and K) Percentages of different mutation types (J) and genotypes (K) for individual blastomeres of 8- to 16-cell embryos with Tyr targeting. LED, large-fragment exon deletion; Bi-allelic, bi-allelic mutations; Mono-allelic, mono-allelic mutations. Number, total alleles, or blastomeres analyzed. (L) Percentages of different genotypes of entire 8- to 16-cell embryos with Tyr targeting. Number, total embryos analyzed.
DNA genotyping analysis of the Tyr gene-edited embryos (n = 12) and mice (n = 6) created by 4-sgRNA targeting (sgRNA-Tyr-B+C+D+E) confirmed Tyr complete knockout. It also revealed that with an increasing number of sgRNAs used, the percentage of gene-edited mice exhibiting large-fragment exon deletion (defined as > 30 bp in this work) rather than regular indels (defined as ≤ 30 bp)11 at the target locus gradually increased (Figure 2F-2I; Supplementary information, Figure S1A-S1E). We further examined the gene-edited 8- to 16-cell embryos at the single-cell level. We found that all blastomeres created by 3- to 4-sgRNA targeting (n = 116) carried bi-allelic mutations, resulting in complete Tyr knockout in all embryos (n = 21) (Figure 2J-2L; Supplementary information, Figure S1F and S1G). By contrast, blastomeres with single-sgRNA targeting (n = 58) showed 10.3% of WT alleles and 3.4% of mono-allelic mutations, resulting in 67% of embryos (6/9) with Tyr incomplete knockout (Figure 2J-2L; Supplementary information, Figure S1F-S1H). Together, these results indicate that a single allele or two alleles of gene could be completely knocked out in the embryo by a cocktail of targeting sgRNAs in the CRISPR/Cas9 system (C-CRISPR hereafter), when a single exon is targeted with two or more closely spaced sgRNAs. Such high efficiency in complete gene knockout appears to be caused by simultaneous frame-shift mutation and exon deletion.
One-step generation of complete triple knockout
To test whether multiple genes could be completely knocked out in one step by C-CRIPSR injection of the zygotes, we simultaneously targeted Tet1, Tet2, and Tet3, three functionally redundant genes involved in DNA oxidation12 (Figure 3A). After embryo implantation, we harvested E7.5 embryos and examined the presence of Tet genes by assaying 5-hydroxymethylcytosine (5hmC) level, the main product derived from oxidation of 5-methylcytosine (5mC) by Tet proteins. In Tet gene-edited embryos (Group I: Tet1 and Tet2 double targeting by 6 sgRNAs, Tet1, 2-A+B+C; Group III: Tet1, Tet2, and Tet3 triple targeting by 9 sgRNAs, Tet1, 2, 3-A+B+C), about half had degenerated by E7.5 (Figure 3B), consistent with previous reports13,14,15,16. Immunostaining of epiblast tissue sections for 5hmC and 5mC in E7.5 embryos showed a high level of 5hmC in all cells of WT embryos (Figure 3C). By contrast, embryos with Tet1, Tet2, and Tet3 triple targeting by 3 sgRNAs (Group II: one sgRNA for each gene, Tet1, 2, 3-A) showed a mosaic pattern of 5hmC staining, with high level of 5hmC in some cells (Figure 3C). In addition, embryos with Tet1 and Tet2 double targeting by 6 sgRNAs (3 sgRNAs for each gene) showed similar mosaic staining of 5hmC. However, embryos with Tet1, Tet2, and Tet3 triple targeting by 9 sgRNAs (3 sgRNAs for each gene) showed complete absence of 5hmC (Figure 3C), indicating complete deletion of all three Tet genes. The average ratio of 5hmC/5mC was quantified by measuring the intensity of immunofluorescence, and the results showed that the ratio dropped in half in Group I & II and to much lower levels in Group III (Figure 3D). Finally, double-Tet-knockout embryos created with 6 sgRNAs showed reduced birth rate, and triple Tet-knockout embryos created by 9 sgRNAs yielded no live birth (Figure 3E), consistent with previous studies13,14,15,16. These results demonstrate the efficiency of one-step deletion of multiple genes using the C-CRISPR method.

One-step generation of mice with complete triple knockout of Tet genes by C-CRISPR. (A) Schematic diagram of sgRNA-targeting sites at Tet1, Tet2 and Tet3 loci, with three sgRNAs for each locus. (B) Percentages of control and Tet gene-edited E7.5 embryos that were intact. Number, total number of embryos counted (***P < 0.001, **P < 0.01, χ2 test). (C) Immunostaining of tissue sections showing the level of 5hmC (red) and 5mC (green) in E7.5 embryos with different forms of Tet gene targeting. White-dashed line, epiblast of E7.5 embryos, with boxed regions shown at a higher resolution on the right. “5hmC”, 5-hydroxymethylcytosine; “5mC”, 5-methylcytosine; scale bar, 50 μm. (D) The relative levels of 5mC and 5hmC in E7.5 embryos. Each data point represents the ratio (5hmC/5mC) of the average immunofluorescence intensities measured from one tissue section. Error bars, SEM (***P < 0.001, **P < 0.01, unpaired t-test). (E) Birth rate of mice for control embryos and different forms of Tet gene targeting. Number: total number of embryos transferred to surrogates (***P < 0.001, χ2 test).
Phenotype analysis of F0 mice with complete gene knockout by C-CRISPR
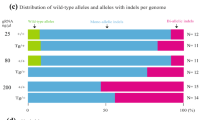
We next employed the C-CRISPR method for phenotypic analysis of the function of various Y-chromosome genes, and tested the efficiency of this one-step complete gene knockout in mice for rapid functional screening of a large number of genes. The Y chromosome is highly specialized for male sex differentiation and fertility17,18. However, so far only a few genes have been assessed for biological function in targeted knockout mice. We decided to target eight individual single-copy Y chromosome genes (Figure 4A). We found that embryos targeted at Erdr1, located in both Y and X chromosomes, resulted in embryonic lethality (Figure 4B). Deletion of the other seven genes individually had no apparent effect on the normal birth rate. Genotyping these mice confirmed the complete deletion of the individual genes (Figure 4C; Supplementary information, Table S1). Consistent with previous reports19,20,21, deletion of Sry resulted in sex reversal of male embryos (Figure 4D) and male mice targeted for Eif2s3y deletion were infertile and had hypoplastic testes (Figure 4E-4H). By contrast, all male mice with deletions for each of the other five genes (Zfy1, Ube1y1, Kdm5d, Ddx3y, Usp9y), and their F1 male offspring, were fertile and showed normal reproductive ability (Figure 4E-4I). Taken together, these results showed that the C-CRISPR method could be applied for the rapid phenotypic analysis of gene functions by generating complete gene knockout mice in F0.

Phenotype analysis of F0 mice with individual Y chromosome gene deletion by C-CRISPR. (A) Schematic diagram of eight targeted genes in Y chromosome. Two sgRNAs were designed to target each gene. Red, gene previously studied with conventional knockout mice; green, gene located in both X and Y chromosomes. (B) Birth rate of mice with embryos edited by the C-CRISPR method for deletion of individual Y-chromosome gene. Number, total embryos transferred. Erdr1 targeting resulted in embryonic lethality. (C) Sex ratio of mice with deletion of different Y-chromosome genes. (D) DNA sequence analysis of gene-edited mice in Y chromosome by C-CRISPR. Tails of mice resulting from different gene targeting in Y chromosome were genotyped. LED, large-fragment exon deletion; Indels, insertion, or deletion of bases. Number above each column, total TA clones sequenced. (E-G) Sperm concentration (E) weight-to-body ratio of testis (F) and percentages of sperm with progressive motility (G) for mice with deletion of different Y-chromosome genes. Number, total number of samples counted (***P < 0.001, **P < 0.01, *P < 0.05, χ2 test). (H) Histological analysis of the testis sections from adult mice with deletion of different Y-chromosome genes. Arrow, abnormal vacuoles of seminiferous tubule; arrowhead, sperm. Abnormality was found only in the Eif2s3y deleted testis. Bar, 20 μm. (I) Reproductivity of F0 and F1 mice with deletion of different Y-chromosome genes. Number, total pups obtained.
C-CRISPR-mediated complete gene knockout in monkeys
We next examined whether the C-CRISPR method could be used to generate monkeys with complete gene knockouts. We performed targeted deletion of Prrt2 (mutations in paroxysmal kinesigenic dyskinesia), and Arntl (a core component of the circadian clock) in the cynomolgus monkey (Macaca fascicularis) (Figure 5A). After pretesting the DNA cleavage efficiencies of each sgRNA in monkey embryos or monkey COS-7 cells (Supplementary information, Figure S2A and data not shown), we targeted monkey embryos with 3 sgRNAs of Prrt2 having relatively higher cutting efficiencies (sgRNA-Prrt2-B+C+D). When all blastomeres of each individually injected 8-cell embryos were analyzed, we found that, with the exception of one blastomere that carried mono-allelic mutation, all blastomeres with detectable PCR signals (n = 33, from 11 embryos) carried bi-allelic mutations in Prrt2 gene, resulting in 10 complete knockout embryos out of 11 injected embryos (Figure 5B and 5D, Supplementary information, Figure S2B). By contrast, control 8-cell blastomeres from 8 embryos injected with a single sgRNA (sgRNA-Prrt2-C) showed 90% of WT alleles and 10% of mono-allelic mutations, resulting in 3 incomplete knockout embryos and 5 WT embryos (Figure 5B; Supplementary information, Figure S2B and S2C). For Arntl targeting, we found that all eight examined embryos targeted with 3 sgRNAs (sgRNA-Arntl-A+B+C) showed complete knockout of Arntl (Figure 5C and 5D; Supplementary information, Figure S2D).

One-step generation of complete knockout monkey by C-CRISPR. (A) Schematic diagram of sgRNA-targeting sites at Prrt2 and Arntl loci in monkey. (B and C) Percentages of different mutation types for 8-cell embryos with Prrt2 (B) and Arntl (C) targeting. LED, large-fragment exon deletion. Indels, insertion, or deletion of bases. Number above each column, total alleles analyzed from blastomeres of 8-cell embryos with Prrt2 targeting or total TA clones sequenced from 8-cell embryos with Arntl targeting. (D) Genotyping analysis on single blastomeres of 8-cell embryos targeted with sgRNA-Prrt2-B+C+D or whole 8-cell embryos targeted with sgRNA-Arntl-A+B+C. The sgRNA targeted sequences are labeled in green and the PAM sequences are labeled in red; deleted nucleotides are indicated by hyphens. Dashed lines mark the region omitted for clarity. (E) Photograph of Prrt2-knockout monkey (#11). (F) Percentages of different mutation types found in tail, ear, and blood cells from aborted and live monkeys by Prrt2 targeting. Aborted monkeys with sgRNA-Prrt2-A targeting: monkey #2, #3; live monkeys with sgRNA-Prrt2-A targeting: monkey #4, #8, #10; live monkeys with sgRNA-Prrt2-B+C+D targeting: monkey #11, #12. AA/GC, a point mutation in Prrt2(335N to 335A). (G) Representative sequences from ear, tail, and blood cells of monkey #11 and #12 with sgRNA-Prrt2-B+C+D targeting. The sgRNA-targeting sequences are labeled in green and PAM sequences are labeled in red; deleted nucleotides are indicated by hyphens. Dashed lines mark the region omitted for clarity. (H) Single-cell analysis on blood cells and fibroblasts from two live monkeys with Prrt2 editing (#11, #12). Number, total number of cells analyzed.
To generate Prrt2 knockout monkeys, we transferred 29 embryos targeted with 3 sgRNAs (sgRNA-Prrt2-B+C+D) into nine surrogates and obtained one pregnant monkey, which gave birth of two live male monkeys (Figure 5E). Further DNA sequencing analysis of the ear and tail tissues as well as blood cells from these two monkeys showed that one monkey (#11) carried complete Prrt2 knockout and the other (#12) an incomplete knockout of Prrt2, with a large-fragment exon deletion and point mutations (335N to 335A) (Figure 5F to 5H; Supplementary information, Figure S2E to S2G). By contrast, after 55 embryos injected with a single sgRNA (sgRNA-Prrt2-A) were transferred, we obtained six stillborns and four live monkeys. Two of the stillborns (#2 and #3) and three live monkeys (#4, #8 and #10) carried varying extents (20%-60%) of deletion of Prrt2 and the remaining five monkeys exhibited no gene editing (Figure 5F). These results suggest the C-CRISPR method could achieve essentially complete gene knockout in monkeys.
Off-target effect in complete knockout animals by C-CRISPR
Finally, we examined whether multiple-sgRNA targeting by the C-CRISPR method induces more off-target effects than single-sgRNA targeting. As previously described22, we examined up to 10 off-target sites for each sgRNA in six mice derived from a 4-sgRNA-targeting mutagenesis (Tyr-B+C+D+E-#1, #2, #3, #4, #5, #6), and two monkeys from 3-sgRNA targeting (Prrt2-B+C+D-#11, #12). DNA sequencing of the PCR products amplified from these genomic sites showed that no mutations occurred at any of these loci (Supplementary information, Figure S3A and S3B). We also performed whole-genome sequencing on these samples, as well as on two control monkeys from single-sgRNA targeting (Prrt2-A-#8, #9) at a sufficient depth to detect off-target mutations (15 × for the mice and 25 × for the monkeys). We then searched for off-target effects in the genome allowing up to five mismatches of 4 sgRNAs in the mice (Tyr-B, C, D, and E) and 3 sgRNAs (Prrt2-B, C, and D) in the monkeys (Table 1; Supplementary information, Table S2 and S3). Of 10 201 and 19 447 possible off-target sites in mice and monkeys, respectively, we found no indels in the six mice and monkey #12 after filtering variants shared in individual samples as done in previous studies23,24 (Table 1; Supplementary information, Table S3). For monkey #11, we observed indels in three off-target sites, with five mismatches in both PAM-distal and PAM-proximal regions (Supplementary information, Table S3). We further examined the three off-target sites in monkey #11 by PCR amplification and sequencing, and found that one site was not a true off-target site (Supplementary information, Figure S3C and S3D) and the other two sites were associated with a repeated DNA sequence, which could not be distinguished by sequencing (Supplementary information, Table S3). We also examined genomic rearrangements, including deletions, duplications, inversions, and copy number variations, using the similar strategies, and found no rearrangements in the six mice and two monkeys (#11 and #12) (Supplementary information, Table S3). Thus C-CRISPR approach did not induce significant off-target alterations in gene-edited mice and monkeys beyond that expected for CRISPR/Cas9-mediated editing in general1,24,25,26.
Discussion
The CRISPR/Cas9 system is an efficient gene-editing method, but the majority of gene-edited animals show mosaicism that impedes effective phenotypic analysis of the consequence of gene deletion. In this study, we found that a single gene or multiple genes could be efficiently deleted in all cells of nearly 100% of CRISPR/Cas9-injected mice and monkeys, using a modified “C-CRISPR” approach. Such high efficiency in complete gene knockout appears to be caused by simultaneous frame-shift mutation and exon deletion.
There are several distinct features of the C-CRISPR method that ensure efficient gene deletion. First, when using multiple sgRNAs to target a single exon of a gene for which there is only one allele, such as genes in Y chromosome, 2 to 3 sgRNAs are required; for bi-allelic autosomal genes, 4 to 6 sgRNAs are required. Second, the close apposition of DNA exon sites targeted by multiple sgRNAs is a key feature of our approach. Third, it is important to target a key exon of the ORF of the gene27 because the change of only one or two amino acids in a key domain of a protein may completely disrupt its function. Finally, we noted the importance of pretesting the efficiency of sgRNAs in cleaving DNAs in embryos, especially for gene editing in the monkey because the majority of sgRNAs were not efficient. Together, these features promote large-fragment exon deletion in addition to regular indels, leading to complete gene knockout. We summarized a protocol for the application of C-CRISPR in mice and monkeys (Supplementary information, Data S1).
We also performed comprehensive off-target analysis on the animals produced by multiple-sgRNA targeting and found no obvious off-target effects. This is consistent with previous studies that off-target mutations are rare in Cas9-modifed animals produced by dual-sgRNA targeting23. Our selection of sgRNAs with minimal off-target effects, based on the online software (http://crispr.mit.edu/ and http://www.rgenome.net/cas-offinder) could have contributed to the decrease of off-target mutations.
Recently, other strategies have been used to reduce mosaic mutations in the mouse and monkey28,29. By electroporation of Cas9 protein/sgRNA into in vitro fertilized zygotes at the early pronuclear stage, Hashimoto et al. generated non-mosaic mutants at two gene loci in the mouse28. However, more gene loci need to be tested and whether this approach also works in other species, such as non-human primates, needs to be proven, since the DNA cleavage efficiency of sgRNA is much lower in monkey embryos than in mouse embryos2. Tu et al. reported that promoting Cas9 degradation could also reduce mosaic mutations in non-human primate embryos, resulting in 28.97% monkey embryos with mono-allelic or bi-allelic mutations in all cells at one gene locus29. In our study, we have shown that the C-CRISPR approach produces complete gene knockout monkey embryos with much higher efficiency (100% for Arntl and 91% for Prrt2 achieving bi-allelic mutations in all cells). Our approach with such high efficiency could markedly reduce the number of animals needed for experiments. This is highly desirable from the ethical viewpoint, especially when using non-human primates as animal models. Meanwhile, it would be interesting to combine our multiple sgRNAs strategy with Cas9 protein in early pronuclear zygotes or modified ubiquitin-Cas9 mRNA to generate animals carrying the same genetic mutation in future studies.
The C-CRISPR approach can be particularly useful for large-scale (10-100 genes) functional screening in F0 mice. For example, a large number (> 20) of single-gene knockout F0 mice could be obtained by a single round of injection of ∼ 100 zygotes and three rounds of such injections may be carried out in a day by one person. Another application of this approach in mice is to generate complete gene-knockout blastocysts for the purpose of interspecies blastocyst complementation, which is at present not feasible but highly desirable30. Finally, the C-CRISPR approach also offers a more efficient one-step process for studying gene function in monkey and human embryos. In this study, we were able to obtain one complete gene knockout monkey out of two live monkeys produced by this method, whereas no complete knockout monkey was obtained out of 10 monkeys produced by the single-sgRNA-targeting method. Furthermore, our monkey embryo data also indicate that this approach will make genetic studies in monkeys much more realistic in the future.
Materials and Methods
Animal ethics statement
The use and care of animals complied with the guideline of the Biomedical Research Ethics Committee at the Shanghai Institutes for Biological Science, Chinese Academy of Science (CAS), which approved the application entitled “Reproductive physiology of cynomolgus monkey and establishment transgenic monkey” (#ER-SIBS-221106P).
Production of Cas9 mRNA and sgRNA
The T7 promoter was added to the Cas9 coding region by PCR amplification of px260, using primer Cas9 F and R (Supplementary information, Table S4). The T7-Cas9 PCR product was purified and used as the template for in vitro transcription (IVT) using mMESSAGE mMACHINE T7 ULTRA kit (Life Technologies). The T7 promoter was added to the sgRNA template by PCR amplification of px330, using primers listed in (Supplementary information, Table S4). The T7-sgRNA PCR product was purified and used as the template for IVT using MEGA shortscript T7 kit (Life Technologies). Both the Cas9 mRNA and the sgRNAs were purified using the MEGA clear kit (Life Technologies) and eluted in RNase-free water.
Zygote injection, embryo culturing, and embryo transplantation
For mice gene editing, super ovulated female C57BL/6 mice (3 weeks old) or B6D2F1 (C57BL/6 × DBA2J) mice (7-8 weeks old) were mated to C57BL/6 or B6D2F1 males, and fertilized embryos were collected from oviducts. For GFP targeting, zygotes were harvested after mating homozygous Actin-GFP transgenic31 male mice with WT female mice. Cas9 mRNA (50 ng/μl) and sgRNAs (50 ng/μl for each sgRNA in most of experiments and 20 ng/μl for each sgRNA in Tet1, 2, 3 triple-gene knockout experiments) were mixed and injected into the cytoplasm of fertilized eggs with well recognized pronuclei in a droplet of HEPES-CZB medium containing 5 μg/ml cytochalasin B (CB) using a FemtoJet microinjector (Eppendorf) with constant flow settings. The injected zygotes were cultured in KSOM medium with amino acids at 37 °C under 5% CO2 in air until the 2-cell stage by 1.5 days. Thereafter, 25-30 2-cell embryos were transferred into oviducts of pseudopregnant ICR females at 0.5 dpc.
For monkey gene editing, laparoscopy was used for oocyte collection. Oocytes were aspirated from follicles 2-8 mm in diameter, about 32-36 h after hCG stimulation32. The collected oocytes were cultured in the pre-equilibrated maturation medium. Metaphase II arrested oocytes were used to perform intracytoplasmic sperm injection, and fertilization was confirmed by the presence of two pronuclei. The zygotes were injected with Cas9 mRNA (100 ng/μl) and sgRNAs (50 ng/μl for each sgRNA). After injection, the embryos were cultured in HECM-9 medium until they were transferred to surrogates on the next day. Some embryos were cultured until the morula/blastocyst stage and harvested for genome extraction and analysis.
Single-cell PCR analysis
For picking up and transferring single cells, we used a glass capillary under a dissection microscope. Mouse or monkey 8-16 cell stage embryos were digested with acid Tyrode solution to remove the zona pellucida. Then the embryos were transferred into 0.25% trypsin and the individual blastomeres were separated by gentle pipetting. Finally, the blastomeres were washed in 0.25% trypsin for 7 to 10 times and transferred into a PCR tube. Fibroblast or white blood cells were then diluted stepwise with KSOM until the cells were fully dispersed. Following 7-10 washes with KSOM, a single cell was transferred into a PCR tube. An aliquot of 1.5 μl lysis buffer containing 0.1% tween 20, 0.1% Triton X-100 and 4 μg/ml proteinase K was then pipetted into the tube. Each tube was centrifuged to facilitate mixing. The lysis was performed at a temperature of 56 °C for 30 min, followed by 95 °C for 5 min. The products of the lysis program were used as templates in a nested PCR analysis. All operations were carried with care to prevent contaminating the samples.
Genotyping
The genotypes of mutant mice were determined by PCR of genomic DNA extracted from tails. ExTaq was activated at 95 °C for 3 min, and PCR was performed for 34 cycles at 95 °C for 30 s, 62 °C for 30 s, and 72 °C for 1 min, with a final extension at 72 °C for 5 min. To amplify DNA from blastocysts, single blastocysts were washed six times with KSOM before they were transferred directly into PCR tubes containing 1.5 μl embryo lysis buffer (0.1% tween 20, 0.1% Triton X-100 and 4 μg/ml proteinase K) and incubated at 56 °C for 30 min, heat inactivated with proteinase K at 95 °C for 10 min. PCR amplification was performed using nested primer sets. ExTaq was activated at 95 °C for 3 min, and PCR was performed for 34 cycles at 95 °C for 30 s, 62 °C for 30 s, and 72 °C for 1 min, with a final extension at 72 °C for 5 min. Secondary PCR was performed using 0.5 μg primary PCR product and nested inner primer. PCR was carried out in the same reaction mixture. The PCR product was gel purified and cloned using a pMD-19t cloning kit (Takara) following the manufacturer's instructions. Colonies were picked from each transformation and then Sanger sequencing was applied to detect mutations.
HE staining and immunostaining
To analyse testis, part of the tissue was fixed in Bouin's solution overnight. The fixed tissues were then embedded into paraffin, and 5-m sections were cut with a Microtome. After being deparaffinized, and rehydrated, sections were stained with hematoxylin-eosin. For 5hmC staining, post-implantation embryos were dissected from pregnant females at the day indicated. Then embryos were fixed with 4% paraformaldehyde, embedded in OCT compound (Sakura) and 8 μm sections were cut. After washing with PBS, the sections were treated with hydrochloric acid solution (4N hydrochloric acid, 0.1% Triton X-100 in distilled water), followed by washing with PBS and permeabilization with blocking buffer (1% BSA, 10% goat serum, PBS containing 0.3% Triton X-100). Next, sections were incubated with primary antibodies (mouse anti-5mC antibody, 1:500, Eurogentec#BI-MECY-0100; rabbit anti-5hmC antibody, 1:1 000, Active Motif #39792) overnight at 4 °C and secondary antibodies at room temperature for 1 h. Finally, the slides were mounted in Anti-fade Reagent (Invitrogen) and imaged using a LEICA TCS SP5 II confocal microscope. The signal intensity was determined using Leica Application Suite software. Note, for embryos with Tet1, 2, 3 triple KO, we used the background (non-specific) for 5hmC to calculate the average ratio of 5hmC/5mC.
Computer-assisted sperm analysis
This analysis was carried out as described in a previous study33. Sperm were diluted to 3-6 × 106 spermatozoa/ml in a buffered solution (NaCl 7.0 g/l, KCl 149 mg/l, CaCl2.2H2O 147 mg/l, NaHCO3 2.1 g/l, MgSO4.7H2O 296 mg/l, NaH2PO4.H2O 49.7 mg/l, Glucose 1 g/l, Na pyruvate 121 mg/l, Sucrose 6.63 g/l, TAPSO (buffer) 2.59 g/l (10 mM), Penicillin 57 mg/l, Streptomycin 29 mg/l, pH = 7.3∼7.4) and warmed on a 37 °C plate for 20 min. Sperm suspensions were gently mixed before measuring motility. For each motility measurement, a 5 μl aliquot of sperm suspension was loaded by capillary action using a large-bore pipette tip into the counting chamber (depth, 10 μm) of a pre-warmed Leja slide and analyzed using a computer-assisted semen analysis machine (HTM-TOX IVOS sperm motility analyzer, Animal Motility, version 12.3A; Hamilton Thorne Research). The magnification was 10×. All samples were analyzed at least twice to discard errors due to incorrect sampling. At least five fields per sample were acquired, recording at least 100 motile sperm. Image sequences were saved and subsequently analyzed.
Whole-genome sequencing and off-target analysis
Whole-genome sequencing was carried out using the Illumina HiSeq X Ten at mean coverages of 15× (sgRNA-Tyr-B+C+D+E-#1, #2, #3, #4, #5, #6) and 25× (sgRNA-Prrt2-B+C+D-#11, #12). Qualified reads were mapped to the mouse reference genome (mm10) and the assembly Macaca fascicularis genome (v5) by speedseq34 (https://github.com/hall-lab/speedseq) with default parameters. All of the mapped data is available from the SRA under accession SRP092889. Potential off-target sites of sgRNAs were predicted as previously reported35 (http://www.rgenome.net/cas-offinder/). We extracted all the off-target sites with no more than 5 mismatches for each sgRNA. FreeBayes36 (v0.9.10) and LUMPY37 (https://github.com/arq5x/lumpy-sv) were run on the aligned sequence files (BAM files) for short indel detection and structural variation discovery. Next, we downsized the short indel variations to the predicted off-target sites, these short indels were then filtered to remove variations overlapped in our samples and the remaining variations were manually inspected by mapping sequencing reads in these regions to the extracted reference sequences with BLAST for potential indel patterns. Finally, variants associated with repetitive DNA sequence were also discarded as these cannot be distinguished from the large number of naturally occurring variants in repetitive DNA sequence in individual animals. For the structural variations, the same strategies were used except that the searching regions were broadened to the 250bp up- and down-stream of the potential off-target sites. Variations after each filtering step were listed in the Supplementary information, Table S3.
Statistics
P values were determined by Student's t-test or χ2 test for all measurements. All error bars denote SEM.
Author Contributions
EZ designed and performed mouse experiments. YW and DL performed mouse embryo injection. B-AW and LS performed Tet1, 2, 3 triple knockout experiments. Y-JC and ZL performed monkey Prrt2 gene knockout experiments. KL performed monkey behave test. Y-JC and J-WL performed Prrt2 gene knock out monkey genotyping analysis. ZL, YW, Y-HN, and C-CZ performed monkey oocytes collection and embryos transfer experiments. WW and XH performed HE staining and reproductive analysis. XH, XY, and XW performed mice mating and genotyping. WY and QW transferred embryos. YS performed data analysis on off-target effects. G-LX helped with the design of Tet gene deletion experiments and provided comments on the manuscript. JL helped with the design of GFP and Tyr gene deletion experiments. ZX and QS designed the experiments of Prrt2 knockout in monkey. HY supervised the project, designed experiments and wrote the paper with other helps.
Competing Financial Interests
The authors declare no competing financial interests.
References
Wang H, Yang H, Shivalila CS, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013; 153:910–918.
Niu Y, Shen B, Cui Y, et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 2014; 156:836–843.
Yen ST, Zhang M, Deng JM, et al. Somatic mosaicism and allele complexity induced by CRISPR/Cas9 RNA injections in mouse zygotes. Dev Biol 2014; 393:3–9.
Izpisua Belmonte JC, Callaway EM, Caddick SJ, et al. Brains, genes, and primates. Neuron 2015; 86:617–631.
Jennings CG, Landman R, Zhou Y, et al. Opportunities and challenges in modeling human brain disorders in transgenic primates. Nat Neurosci 2016; 19:1123–1130.
Sato M, Koriyama M, Watanabe S, et al. Direct injection of CRISPR/Cas9-related mRNA into cytoplasm of parthenogenetically activated porcine oocytes causes frequent mosaicism for indel mutations. Int J Mol Sci 2015; 16:17838–17856.
Kotani H, Taimatsu K, Ohga R, Ota S, Kawahara A . Efficient multiple genome modifications induced by the crRNAs, tracrRNA and Cas9 protein complex in zebrafish. PloS ONE 2015; 10:e0128319.
Sung YH, Kim JM, Kim HT, et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Res 2014; 24:125–131.
Chen Y, Zheng Y, Kang Y, et al. Functional disruption of the dystrophin gene in Rhesus monkey using CRISPR/Cas9. Hum Mol Genet 2015; 24:3764–3774.
Zhou J, Wang J, Shen B, et al. Dual sgRNAs facilitate CRISPR/Cas9-mediated mouse genome targeting. FEBS J 2014; 281:1717–1725.
Paquet D, Kwart D, Chen A, et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016; 533:125–129.
Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y . Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010; 466:1129–1133.
Gu TP, Guo F, Yang H, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011; 477:606–610.
Kang J, Lienhard M, Pastor WA, et al. Simultaneous deletion of the methylcytosine oxidases Tet1 and Tet3 increases transcriptome variability in early embryogenesis. Proc Natl Acad Sci USA 2015; 112:E4236–4245.
Dawlaty MM, Breiling A, Le T, et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell 2013; 24:310–323.
Dai HQ, Wang BA, Yang L, et al. TET-mediated DNA demethylation controls gastrulation by regulating Lefty-Nodal signalling. Nature 2016; 538:528–532.
Zhou X, Xin J, Fan N, et al. Generation of CRISPR/Cas9-mediated gene-targeted pigs via somatic cell nuclear transfer. Cell Mol Life Sci 2015; 72:1175–1184.
Tang L, Gonzalez R, Dobrinski I . Germline modification of domestic animals. Anim Reprod 2015; 12:93–104.
Berta P, Hawkins JR, Sinclair AH, et al. Genetic evidence equating SRY and the testis-determining factor. Nature 1990; 348:448–450.
Kato T, Miyata K, Sonobe M, et al. Production of Sry knockout mouse using TALEN via oocyte injection. Sci Rep 2013; 3:3136.
Matsubara Y, Kato T, Kashimada K, et al. TALEN-mediated gene disruption on Y chromosome reveals critical role of EIF2S3Y in mouse spermatogenesis. Stem Cell Dev 2015; 24:1164–1170.
Hsu PD, Scott DA, Weinstein JA, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 2013; 31:827–832.
Iyer V, Shen B, Zhang W, et al. Off-target mutations are rare in Cas9-modified mice. Nat Methods 2015; 12:479.
Yang L, Grishin D, Wang G, et al. Targeted and genome-wide sequencing reveal single nucleotide variations impacting specificity of Cas9 in human stem cells. Nat Commun 2014; 5:5507.
Veres A, Gosis BS, Ding Q, et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell 2014; 15:27–30.
Li W, Teng F, Li T, Zhou Q . Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol 2013; 31:684–686.
Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR . Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol 2015; 33:661–667.
Hashimoto M, Yamashita Y, Takemoto T . Electroporation of Cas9 protein/sgRNA into early pronuclear zygotes generates non-mosaic mutants in the mouse. Dev Biol 2016; 418:1–9.
Tu Z, Yang W, Yan S, et al. Promoting Cas9 degradation reduces mosaic mutations in non-human primate embryos. Sci Rep 2017; 7:42081.
Wu J, Platero-Luengo A, Sakurai M, et al. Interspecies chimerism with mammalian pluripotent stem cells. Cell 2017; 168:473–486 e415.
Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y . 'Green mice' as a source of ubiquitous green cells. FEBS Lett 1997; 407:313–319.
Sun Q, Dong J, Yang W, et al. Efficient reproduction of cynomolgus monkey using pronuclear embryo transfer technique. Proc Natl Acad Sci USA 2008; 105:12956–12960.
Goyal HO, Braden TD, Mansour M, Williams CS, Kamaleldin A, Srivastava KK . Diethylstilbestrol-treated adult rats with altered epididymal sperm numbers and sperm motility parameters, but without alterations in sperm production and sperm morphology. Biol Reprod 2001; 64:927–934.
Chiang C, Layer RM, Faust GG, et al. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods 2015; 12:966–968.
Bae S, Park J, Kim JS . Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014; 30:1473–1475.
Watson SJ, Welkers MR, Depledge DP, et al. Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos Trans R Soc Lond B Biol Sci 2013; 368:20120205.
Layer RM, Chiang C, Quinlan AR, Hall IM . LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 2014; 15:R84.
Acknowledgements
We thank Drs Mu-ming Poo and Dangsheng Li for comments on the manuscript. This work was supported by CAS Strategic Priority Research Program (XDB02050007, XDA01010409), the National High-tech R&D Program (863 Program; 2015AA020307), the National Natural Science Foundation of China grants (31522037, 31500825), China Youth Thousand Talents Program (to HY), Break-through project of Chinese Academy of Sciences, Shanghai Sailing Plan for the Young Scientific Talents (15YF1414700), The National Key Technology R&D Program of China (2014BAI03B00, QS), Shanghai City Committee of science and technology project (16JC1420202, HY; 14140900100, Q.S.), CAS Hundreds of Talents Program of China (QS).
Author information
Authors and Affiliations
Corresponding authors
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Genotyping analysis of gene-edited mice by C-CRISPR. (PDF 707 kb)
Supplementary information, Figure S2
Genotyping analysis of genegene-edited edited edited monkey embryos and tissues by C-CRISPR. (PDF 297 kb)
Supplementary information, Figure S3
Off-target analysis of mice resulting from Tyr targeting and monkeys resulting from Prrt2 targeting. (PDF 836 kb)
Supplementary information, Table S1
Phenotypic analysis of F0 mice with Y-chromosome gene deletions generated by C-CRISPR (PDF 160 kb)
Supplementary information, Table S2
Summary of HiSeq X Ten sequencing (PDF 183 kb)
Supplementary information, Table S3
Summary of genomic rearrangements, off-target effect and filtering steps (XLSX 18 kb)
Supplementary information, Table S4
sgRNA sequences and primers sgRNA sequences (PDF 519 kb)
Supplementary information, Data S1
C-CRISPR-mediated complete gene knockout in mice and monkeys. (PDF 240 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zuo, E., Cai, YJ., Li, K. et al. One-step generation of complete gene knockout mice and monkeys by CRISPR/Cas9-mediated gene editing with multiple sgRNAs. Cell Res 27, 933–945 (2017). https://doi.org/10.1038/cr.2017.81
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2017.81
Keywords
This article is cited by
-
m6A methylation modification and immune infiltration analysis in osteonecrosis of the femoral head
Journal of Orthopaedic Surgery and Research (2024)
-
Identification of oleic acid as an endogenous ligand of GPR3
Cell Research (2024)
-
Efficient prime editing in two-cell mouse embryos using PEmbryo
Nature Biotechnology (2024)
-
Skin graft with dermis and appendages generated in vivo by cell competition
Nature Communications (2024)
-
Whole genome sequencing of mouse lines divergently selected for fatness (FLI) and leanness (FHI) revealed several genetic variants as candidates for novel obesity genes
Genes & Genomics (2024)
