
Abstract
The divergence of archaea, bacteria and eukaryotes was a fundamental step in evolution. One marker of this event is a major difference in membrane lipid chemistry between these kingdoms. Whereas the membranes of bacteria and eukaryotes primarily consist of straight fatty acids ester-bonded to glycerol-3-phosphate, archaeal phospholipids consist of isoprenoid chains ether-bonded to glycerol-1-phosphate. Notably, the mechanisms underlying the biosynthesis of these lipids remain elusive. Here, we report the structure of the CDP-archaeol synthase (CarS) of Aeropyrum pernix (ApCarS) in the CTP- and Mg2+-bound state at a resolution of 2.4 Å. The enzyme comprises a transmembrane domain with five helices and cytoplasmic loops that together form a large charged cavity providing a binding site for CTP. Identification of the binding location of CTP and Mg2+ enabled modeling of the specific lipophilic substrate-binding site, which was supported by site-directed mutagenesis, substrate-binding affinity analyses, and enzyme assays. We propose that archaeol binds within two hydrophobic membrane-embedded grooves formed by the flexible transmembrane helix 5 (TM5), together with TM1 and TM4. Collectively, structural comparisons and analyses, combined with functional studies, not only elucidated the mechanism governing the biosynthesis of phospholipids with ether-bonded isoprenoid chains by CTP transferase, but also provided insights into the evolution of this enzyme superfamily from archaea to bacteria and eukaryotes.
Similar content being viewed by others

Introduction
Phospholipids are the key components of the cell membranes of all living organisms, as they play vital roles in the formation and stabilization of the lipid bilayer, maintain the permeability and fluidity of the barrier1,2,3, and provide an essential compartment for biological activity such as lipid and membrane protein biogenesis, transport, and energy transduction4,5,6. In addition, many types of phospholipids play important regulatory roles in cell signaling, membrane trafficking, apoptosis, and immunity7,8,9.
A key step in membrane phospholipid synthesis is catalyzed exclusively by transmembrane enzymes of the CTP transferase superfamily: transfer of CMP to a glycerol-phosphate backbone, resulting in the formation of CDP-diacylglycerol (in bacteria) or CDP-archaeol (in archaea)1,3,10. Archaea can be distinguished from bacteria by their use of a glycerol-1-phosphate (G1P) backbone, rather than a glycerol-3-phosphate (G3P) backbone, to link isoprenoid hydrocarbon side chains via an ether bond11. Early evolutionary hypotheses proposed that archaea and bacteria diverged directly from a common ancestor (cenancestor) that had a mixed heterochiral membrane12 (Supplementary information, Figure S1). Since the associated “lipid divide” that occurred during the divergence of archaea and bacteria from the cenancestor is considered evolutionarily significant, an intriguing question is how the ether- and ester-based phospholipid biosynthesis pathways evolved in these organisms, respectively. In archaea, members of the membrane-embedded CTP transferase superfamily share considerable sequence similarity (Supplementary information, Figure S2A), whereas those of bacteria and eukaryotes are not well conserved13 (Supplementary information, Figure S2B). Extensive studies have characterized representative CDP-diacylglycerol synthase (CDS) proteins, which are integral membrane enzymes, in Escherichia coli, Saccharomyces cerevisiae, mice, and humans by their preference for activated CTP (deoxy-CTP (dCTP)) or other nucleotides as polar head groups and phosphatidic acid for phospholipid biosynthesis14,15,16,17,18,19. Meanwhile, the first archaeal CDP-archaeol synthase (CarS) was only recently identified and was shown to catalyze an essential step in CDP-archaeol formation, namely, the transfer of CMP to its specific archaeal lipid substrate, archaeol20 (Figure 1A), however, the enzymatic mechanism of CTP transferases remains poorly characterized. Structural and biochemical studies of CarS are, therefore, necessary to reveal how this intramembrane CTP transfer step is catalyzed in membrane bilayers of specific ether lipids and to determine whether these enzymes are structurally related to CDS, most notably CDS from Thermotoga maritima (TmCdsA), the only CDS family member21 using phosphatidic acid as a substrate whose structure has been determined.

ApCarS is a CTP transferase. (A) Cartoon representation of the reaction catalyzed by ApCarS, which generates CDP-archaeol from CTP and DGGGP. The C-labeled hexagon and R-labeled pentagon represent the cytosine and ribose portions of CTP, respectively. The phosphate groups are depicted as red circles, and ApCarS is in green. (B) ITC raw data and binding trace for CTP titrated into a solution containing ApCarS and Mg2+: Kd = 0.20 M; N = 0.36; ΔH = −9.180 kcal/mol; and ΔS = −2.18 cal/mol. See details in Supplementary information, Table S2. Because of endogenously bound CTP that co-purified with the protein, the binding site “N” value is lower than 1. (C) Representative ITC raw data and binding trace for CTP titrated into a solution containing ApCarS in the absence of Mg2+: Kd = 1.67 μM; N = 0.31; ΔH = −1.134 kcal/mol; and ΔS = −11.6 cal/mol. See details in Supplementary information, Table S2. Because of endogenously bound CTP that co-purified with the protein, the binding site “N” value is lower than 1. (D) Graphic depiction of ApCarS activity. The activity of purified ApCarS was assessed with chemically synthesized DGGGP as a substrate at different temperatures in the presence or the absence of 2 mM CTP and in the presence of Mg2+ and EDTA. The production of CDP-archaeol (m/z = 1 020.55 (M-H)) was verified by HPLC-MS analysis (Supplementary information, Figure S3).
Here, we report the crystal structure of CarS from Aeropyrum pernix (ApCarS) in the CTP- and Mg2+-bound state at a resolution of 2.4 Å. This structure reveals that ApCarS exhibits clear cytoplasmic and transmembrane domains. Supported by structural and biochemical evidence, our study provides a structural basis for the binding of Mg2+, CTP, and the isoprenoid chains of ether-bonded lipids to this enzyme. Moreover, our findings suggest that the specific binding of lipophilic substrates drives catalysis.
Results
ApCarS is a CTP transferase
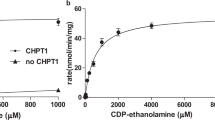
We purified CarS proteins from various archaeal species and obtained X-ray diffracting crystals from Aeropyrum pernix K1 CarS (ApCarS) after detergent screening. ApCarS shares 37% sequence identity with the functionally characterized CarS of Archaeoglobus fulgidus (AfCarS)20. As Mg2+ is essential for the optimal enzymatic activity of AfCarS20, we examined the CTP-binding affinities of ApCarS in the presence or the absence of Mg2+ using isothermal titration calorimetry (ITC). Purified ApCarS exhibited strong CTP-binding activity, with dissociation constants of 0.2 and 1.67 μM in the presence and the absence of Mg2+, respectively (Figure 1B and 1C). We subsequently evaluated the CTP transferase activity of ApCarS using an in vitro catalytic assay. For these analyses, purified ApCarS was incubated with CTP and 2,3-bis-O-geranylgeranyl sn-glycerol-phosphate (DGGGP) at 37 °C for 1 h, and the production of CDP-archaeol was monitored by liquid chromatography-mass spectrometry (LC-MS) (Figure 1D; Supplementary information, Figure S3). As anticipated, CDP-archaeol was only detected in the presence of CTP. ApCarS enzymatic activity was inhibited by EDTA (Figure 1D), which was rescued by the addition of Mg2+ or Mn2+ but not Ca2+ or Zn2+ (Supplementary information, Figure S4A). Interestingly, the Mg2+-dependent activity of ApCarS was enhanced by the addition of K+ or Li+ but not Na+. In the presence of Mg2+ and K+, the Km for CTP during CDP-archaeol formation was 1.28 mM, with a Vmax of 2.1 μmol/min/mg (Supplementary information, Figure S4B). The ApCarS-mediated production of CDP-archaeol was markedly enhanced at higher temperatures, and the highest catalytic activity of ApCarS was observed at 90 °C (Figure 1D). Notably, binding assays were performed at 25 °C, a temperature at which enzymatic activity is minimal, whereas all other enzymatic activity assays were performed at 90 °C. These results are, therefore, consistent with the fact that A. pernix K1 is a thermophilic archaeon that grows at extreme temperatures. Together, our data indicate that ApCarS is a functional homolog of AfCarS.
Structure of CTP-bound ApCarS
We successfully crystallized the CTP-ApCarS complex using the lipid cubic phase (LCP) method and determined its structure at a resolution of 2.4 Å (Supplementary information, Table S1). ApCarS is primarily composed of five transmembrane helices (TMs), with a large charged cavity at the cytoplasmic face (Figure 2A and 2B). The cytoplasmic cavity primarily comprises TM1, TM2, TM3 and TM4, and is loosely occluded by TM5. The remaining portion of the cavity is formed by two cytoplasmic loops (CLs): CL1 (between TM1 and TM2) and CL2 (between TM3 and TM4; Figure 2A and 2B). CL1 and CL2 form the cytoplasmic domain (CPD), which caps the transmembrance domain (TMD). CL1, which is longer than CL2, is stabilized by packing against one side of TM3 (Figure 2A and Supplementary information, Figure S5A).

Structure of the ApCarS-CTP complex. (A) Cartoon of the ApCarS-CTP complex along the membrane plane. The cytoplasmic domain (CPD) and the TMD are indicated with different colors; TM5 is highlighted in yellow. (B) Stick diagram depicting the amino-acid residues within the active site; cytoplasmic loops 1 (CL1) and 2 (CL2) are highlighted in orange and pink, respectively. A 2Fo-Fc omit map of CTP (contoured at 2.2σ) is shown in violet mesh. (C) Trypsin digestion of ApCarS in the presence of CTP and/or Mg2+. Proteolytic fragments were visualized by SDS-PAGE and Coomassie staining. (D) Electrostatic surface representation of ApCarS bound to CTP. CTP is shown as a ball-and-stick representation within the active site. The large central cavity contains a polar pocket (red and blue) and a hydrophobic pocket (gray) for binding to the phosphate groups and the cytosine ring of CTP, respectively.
As defined by unbiased electron density, CTP binds to one side of the central cavity of ApCarS (Figure 2B and Supplementary information, Figure S6A) and stabilizes the CPD through tight contacts. Consistent with structural observations, limited proteolysis assays indicated that purified ApCarS exhibited increased trypsin resistance in the presence of CTP, even in the absence of Mg2+ (Figure 2C). ApCarS-bound CTP adopts a curved conformation with the triphosphate groups nearly perpendicular to the nucleobase. The binding pocket for the phosphate groups of CTP is polarized, with positive and negative charges on the CPD and TMD side, respectively, which potentially fix the phosphate conformation via the pull-push effects of the opposite charges. The β- and γ-phosphate groups of CTP are buried, whereas the α-phosphate group is partially solvent-exposed, presumably for nucleophilic attack by the substrate DGGGP. The nucleobase portion of CTP fits within a hydrophobic pocket located between the CPD and the TMD (Figure 2D).
Recognition of CTP and metal ions
Recognition of CTP by ApCarS occurs through a combination of extensive polar and hydrophobic interactions. The γ-phosphate group of CTP forms salt bridges with Lys107 and Arg108 of ApCarS, and hydrogen bonds with the side chain of Ser104 and the amide nitrogen of Gly115. Additionally, the γ-phosphate group establishes water-mediated hydrogen bonds with the side chains of Asp100, Asp122, and Asp125. Collectively, these interactions result in the complete burial of this phosphate group (Figures 2D and 3A). In contrast, fewer interactions occur between the two other phosphate groups of CTP and ApCarS. O1 of the β-phosphate group and O3′ of the sugar ring form a pair of hydrogen bonds with Arg114, whereas the α-phosphate group forms an indirect interaction with Asn28 and direct interactions with Lys57 and Asp100 (Figure 3A). In addition to a hydrogen bond with Arg114, the sugar ring contributes to CTP binding by being sandwiched between Val32 and Lys57 (Figure 3B). Although Gly56 does not interact with CTP (they are separated by a minimum distance of ∼4.0 Å), this residue might play a role in CTP recognition through steric limitation. Meanwhile, the cytosine of CTP further inserts into the CTP-binding pocket, forming hydrogen bonds with Thr58 and packing against Pro31 and Lys57. An intramolecular hydrogen bond is formed between O1 of the β-phosphate group and O3′ of the sugar ring of CTP, which likely maintains the curved conformation of the ApCarS-bound CTP (Figure 3B). Structure-based sequence alignments showed that most CTP-interacting residues (Asn28, Asp41, Thr58, Asp100, Ser104, Lys107, Arg108 and Asp122) are strictly conserved, although some (Pro31, Val32, Asp55, Lys57 and Asp125) are less conserved among CarS family members. All residues (Asn28, Lys57, Asp100, Thr104, Lys107, Arg108, Asp122 and Asp125) that interact with CTP phosphate groups are charged or polar. In contrast, the nucleobase-interacting residues (Pro31, Val32 and Thr58) are mostly hydrophobic. Collectively, these results suggest that members of the CarS family share a common mechanism for CTP recognition (Supplementary information, Figure S2A).

Recognition of CTP and metal ions. (A) Recognition of a CTP phosphate group. (Left) Network of interactions formed between the γ-phosphate group of CTP and residues within the polar pocket of the ApCarS active site. Amino-acid residues are indicated in ball-and-stick representation, water molecules are indicated by red balls, and CTP is shown as a green stick. (Right) Residues K57, D100 and R114 participate in coordinating CTP within the active site via interactions with oxygen atoms from the α- and β-phosphate, ribose, and the cytosine groups of CTP. K+ is shown as a purple ball. (B) Interactions between CarS and the nucleotide portion of CTP. The blue dashed lines indicate hydrophobic packing. Hydrogen bonds are shown in yellow. (C) The Mg2+ (Mg1) ion in the active site of ApCarS. Mg2+ is bridged with oxygen atoms from the CTP α- and β-phosphate groups, and water molecules. (D) The Mg2+ (Mg2) ion bound at the N-terminal region of ApCarS.
The electron density map of the ApCarS crystal structure shows additional electron density adjacent to the α- and β-phosphate groups (Supplementary information, Figure S6A). A single Mg2+ (Mg1) ion was modeled in the electron density, and it coordinates with the O1 and O2 atoms of the α- and β-phosphate groups as well as three water molecules (Figure 3C and Supplementary information, Figure S6B). A role for Mg2+ in the CTP-binding and catalytic activities of ApCarS is supported by our ITC and enzymatic activity assays (Figure 1B-1D). The importance of Mg2+ in the catalytic activity of ApCarS is reminiscent of the Mg2+-assisted transfer of the β- and γ-phosphate groups of kinase-bound ATP to substrates. However, whereas Mg2+ coordinates with the β- and γ-phosphate groups of kinase-bound ATP molecules, the Mg2+ ion appears to coordinate with the α- and β-phosphate groups of the ApCarS-bound CTP.
Extra density was also observed around the binding site of the CTP α-phosphate group. Modeling a Mg2+ or Li+ ion into this density resulted in large deviations from its optimal coordination distances to the α-phosphate group of CTP, Asn28 and Asp100 of ApCarS and a water molecule. By contrast, reasonable coordination lengths were achieved when a K+ ion was modeled into the density (Figure 3A and Supplementary information, Figure S6C). This assignment is consistent with the structure of the related protein TmCdsA, in which a K+ ion was also detected21, and our biochemical data (Supplementary information, Figure S4A).
Additionally, the center of the periplasmic region also contains a patch of strong electron density. A second Mg2+ (Mg2) was modeled into this region (Figure 3D and Supplementary information, Figure S6D), and it coordinates with the carbonyl oxygen of Arg10, the side chains of Asp8, Asp12 and Glu16, and two water molecules. These Mg2+ (Mg2)-mediated interactions might stabilize the local conformation of ApCarS, allowing the N-terminal portion to fold backward and interact with both TM4 and TM5. The water molecule could then mediate an interaction between Asn82, which is located in the short-turn loop connecting TM2 and TM3, and Tyr139, which in turn packs against Trp9 via a π-π interaction (Supplementary information, Figure S7A). These interactions could act together to maintain the stability of the periplasmic region. Indeed, alanine substitutions of these amino acids greatly reduced the enzymatic activity of ApCarS (Supplementary information, Figure S7B).
To verify the importance of the CTP-interacting residues identified in the crystal structure, we generated a panel of ApCarS variants harboring amino-acid substitutions within the CTP-binding site and evaluated the CTP-binding and CDP-archaeol production activities of these variant proteins (Figure 4; Supplementary information, Table S2 and Figure S8). Consistent with our structural observations, substituting CTP-interacting residues with alanine resulted in a significant reduction or loss of CTP-binding affinity, as indicated by ITC analyses. The catalytic activities of these variant proteins were also significantly compromised. Specifically, mutating Lys57, Asp100, Asp125 and Lys107, which form polar interactions with the phosphate groups of CTP, to alanine abrogated the CTP-binding (Figure 4A) and catalytic activities (Figure 4B) of ApCarS. Similar results were obtained upon mutation of Asn28 (N28A; Figure 4), which interacts with the CTP α-phosphate group in a water- and K+-associated manner (Figure 3A). Notably, although mutating Pro31 (P31A), which packs against the cytosine portion of CTP, disrupted the CTP-binding activity of ApCarS, this variant retained appreciable catalytic activity in the presence of high concentrations of CTP. The precise reason for this discrepancy is unclear, but it might result from residual CTP-binding activity of this mutant that is beyond the detection limit of our assays (Figure 4B).

Characterization of key residues involved in CTP binding and catalysis. (A) Measurement of CTP binding to ApCarS variants by ITC. The indicated residues were mutated to alanine (A), and CTP binding was analyzed in the presence of Mg2+. The representative original ITC data are presented in Supplementary information, Figure S8 and Table S2. (B) CTP transferase activities of ApCarS variants. Residues around the active site were mutated to alanines. n.d., not detected; WT, wild type.
Asp55, located within CL1, does not directly interact with CTP. Instead, this residue forms a bifurcated salt bond with Arg51, thereby stabilizing the conformation of CL1. The D55A variant exhibited moderately reduced CTP-binding activity but strikingly compromised catalytic activity, indicating that a proper CL1 conformation is essential for optimal enzymatic activity of ApCarS (Supplementary information, Figures S5, S8 and Table S2).
ApCarS is a structural homolog of TmCdsA
A Dali search identified the bacterial CDP-DAG synthase TmCdsA (PDB codes: 4q2e and 4q2g)21 as the closest structural homolog of ApCarS, although these two proteins share only 12% sequence identity. The structural homology between the two proteins primarily lies within TM1-TM4 of ApCarS (Figure 5A), suggesting that these domains might share a common evolutionary origin. In particular, key residues surrounding the CTP-binding region of ApCarS are highly conserved in TmCdsA, implying that TmCdsA might use a similar site for CTP binding (Figure 5B). In contrast, the remaining amino-acid sequences of the conserved domains are much less conserved, offering an explanation for the different substrate specificities of these two structurally related enzymes. Furthermore, compared with ApCarS, TmCdsA contains an additional N-terminal TM domain (Figure 5A). Interestingly, this domain is necessary for TmCdsA homodimerization, which is likely important for enzyme activity21.

Structural comparison of ApCarS and TmCdsA. (A) Superimposed structures of ApCarS (gray) and TmCdsA (purple). The TmCdsA (PDB code: 4q2e21) structure showed the highest similarity to that of ApCarS by DALI sever analysis. (B) Comparison of the ApCarS and TmCdsA active sites. Conserved residues are labeled in purple on TmCdsA and in gray on ApCarS. In the secondary sequence alignment of the conservative motifs, X indicates any amino acid. (C) (Left) Docking of the lipid DGGGP into the ApCarS crystal structure (PDB code: 5GUF). Electrostatic potential at the surface of the CTP- and DGGGP-binding sites in ApCarS. (Right) Docking of the lipid DAG into the TmCdsA crystal structure (PDB code: 4Q2E). Electrostatic potential at the surface of the CTP- and DAG-binding sites in TmCdsA. These binding sites are shown in the same orientation based on superimposed structures. Mg2+ and K+ are highlighted as green and purple balls, respectively.
Identification of the putative archaeol-binding site
In ApCarS, TM5 loosely packs against TM1 and TM4, resulting in the formation of two deep grooves adjacent to the CTP-binding site. The amino-acid residues of TM1, TM4, and TM5 that line these two grooves are largely hydrophobic and are conserved among CarS family members (Supplementary information, Figure S2A). Similarly, the structure of TmCdsA also contains a groove, which was proposed to mediate the binding of diacylglycerol (DAG)21. Structural analyses suggested that the two hydrophobic grooves of ApCarS might act as a binding site for the substrate DGGGP. Additionally, a positively charged cavity located adjacent to the α- and β-phosphate groups of CTP (Figure 2D) might be important for the recognition of the negatively charged G1P portion of the substrate. Compared with the N-terminus, the C-terminal portion of TM5 is more flexible, as evidenced by a higher average B-factor (Supplementary information, Figure S10), and might, therefore, play a role in substrate binding and product release.
To support the model proposed above, we conducted modeling studies by docking DGGGP and DAG into the crystal structures of CTP- and Mg2+-bound ApCarS and TmCdsA, respectively (Figure 5C), and then performed a 100-ns molecular dynamics (MD) simulation of the ApCarS model that reached a local equilibrium (Figure 6A). The modeling studies positioned DGGGP into the two lateral grooves (LG): LG1 and LG2, formed by TM5 and TM1 and by TM5 and TM4, respectively (Figure 6A and Supplementary information, Figure S9C), with the G1P group of DGGGP interacting with the charged cavity (Figure 6B and 6C).

Identification of the putative archaeol-binding site. (A) Image depicting the MD model of DGGGP binding to ApCarS in the lipid bilayer. (B) Polar residues adjacent to the active site are involved in binding to the DGGGP head phosphate group, which was verified by enzymatic activity assays as shown in Figure 4B. (C) In vitro lipid-binding assays. Membrane strips containing equal amounts of DGGGP were incubated with purified proteins as indicated (see Materials and Methods). WT, which is known to bind DGGGP, was used as a positive control. Lipids alone were used as negative controls. ApCarS variants were included where indicated. Positive interactions were detected by incubation with anti-6×His antibody. (D) Lateral groove 1 (LG1) is involved in binding one of the lipophilic tails of DGGGP. (Left) Cartoon and electrostatic representation depicting LG1, which is proposed to interact with one tail of DGGGP. (Right) Activity analysis demonstrating that key residues (A29 and L156) are critical for the DGGGP interaction through the LG (Supplementary information, Figure S9C). (E) LG2 is involved in binding the other lipophilic tail of DGGGP. (Left) Cartoon and electrostatic representation depicting LG2, which is proposed to interact with the other DGGGP tail. (Right) Activity analysis demonstrating that key residues (Q123, L124, F126 and H157) are critical for interaction with DGGGP through the LG (Supplementary information, Figure S9C).
To provide experimental evidence that these two grooves comprise a DGGGP-binding site, we first generated a panel of ApCarS variants (Figure 6B) in which the clustered polar residues proposed to bind the polar G1P group were replaced with alanines (N28A, K57A, D100A, D122A, and D125A) and then assayed the DGGGP-binding activity of these variants. Consistent with our hypothesis, the D122A and D125A mutant proteins failed to bind DGGGP, whereas N28A, K57A and D100A showed weak DGGGP-binding activity (Figure 6C). Similar assays were performed by mutating residues (A29W, Q123A, L124W, F126A, L156W, and H157A) that are likely involved in binding the lipophilic tail within the two grooves (Figure 6D and 6E). The A29W, L124W, F126A and L156W variants showed no detectable interaction with DGGGP (Figure 6C). Interestingly, however, the Q123A and H157A variants exhibited enhanced DGGGP-binding activity. Consistent with these data, the catalytic activity of each of these variant proteins, except for Q123A and H157A, was almost completely abrogated (Figure 6B-6E; see Discussion below).
Specific recognition of DGGGP by ApCarS
Our modeling studies also provided clues about the mechanism underlying the specific recognition of DGGGP by ApCarS (Figure 5C). In TmCdsA, the single LG was proposed to recognize the two linear fatty acid chains of ester-bonded lipids (Figure 5C). Compared with the two grooves in ApCarS, this groove is much smaller and likely cannot accommodate the two branched isoprenoid chains of ether-bonded DGGGP. In addition, the extension of lipophilic tails would be restricted by the wall in TmCdsA at one side (Figure 5C). These differences provide an explanation for why TmCdsA is incapable of recognizing DGGGP (Figure 6D and 6E). The distance between the Ala29 and Leu156 side chains in LG1 is ∼6.6 Å (Figures 5C and 6D), sufficient to accommodate a single lipid tail from DGGGP. Consistent with a role for these residues in DGGGP recognition by ApCarS, substitution at these positions with the bulky residue tryptophan, which is expected to narrow LG1, resulted in reduced DGGGP-binding and catalytic activity of ApCarS (Figure 6C and 6D). Similarly, altering LG2, which was modeled to bind the other lipophilic tail of DGGGP, via the amino-acid substitutions (Q123A, L124W or F126A in TM4, or H157A in TM5), resulted in altered DGGGP-binding and catalytic activities of ApCarS (Figure 6C and 6E). The observed inhibitory effect was particularly pronounced for F126A, likely because Phe126 forms hydrophobic interactions with the lipophilic tail of DGGGP (Figure 6E), thus facilitating the correct substrate positioning. Presumably, Gln123 and His157 are also required to orient DGGGP by restricting the lipid tail in the specific groove (Figure 6A). Thus, when these polar residues were substituted with alanine, the flexible hydrophobic tails would be buried deeply in the membrane, and thereby strongly interact with hydrophobic residues and membrane components, likely making it more difficult for DGGGP to reach the CTP molecule for nucleophilic attack (Figure 6E).
Discussion
Here, we report the structural and biochemical characterization of the intramembrane enzyme ApCarS, which is involved in the biosynthesis of archaeal membrane lipids. The protein was co-purified with endogenous CTP from the expression host E. coli, as shown by ITC measurements (Supplementary information, Table S2). This finding was further confirmed by our structural and proteolysis data (Figure 2B and 2C). Enzymatic activity assays demonstrated that ApCarS is a CTP transferase and a functional homolog of AfCarS (Figure 1D). Coupled with biochemical data, our structural and modeling studies provide insights into the mechanisms by which ApCarS recognizes CTP and lipophilic substrates. Notably, the conservation of predicted CTP-interacting and lipid-binding residues indicates that this mechanism is likely conserved among CarS family members (Figure 5B and Supplementary information, Figure S2).
Our data also provide insights into the catalytic mechanism of ApCarS. Specifically, we found that CTP binding stabilizes the CPD of ApCarS (Figure 2C) and this effect is essential for the catalytic activity of this enzyme, as mutations that destabilize CL1 abrogated CTP-binding and catalytic activity (Figure 4 and Supplementary information, Figure S5). Meanwhile, structural comparisons demonstrated that the catalytic core domains of ApCarS and TmCdsA (Figure 5A and 5B; Supplementary information, Figure S9B) are highly conserved; however, conformational differences between ApCarS and TmCdsA were observed (Supplementary information, Figure S9A).
The location of CTP combined with the results of the MD simulations (Figure 6), allows us to propose a mechanism for archaeol modification by ApCarS (Figure 7A). The G1P group of archaeol resides in the polar pocket, where it attacks the CTP α-phosphate group (Figure 6B). Coordination with Mg2+, Asn28, Lys57 and Asp100 renders the α-phosphate group more electrophilic (Figure 3A and 3B), thereby allowing it to react with the negatively charged G1P head of archaeol. The pyrophosphate leaving groups are likely stabilized by the charged residues Lys107, Arg108, Asp122 and Asp125 (Figure 3A). While binding to ApCarS results in burial of the γ- and β-phosphate groups of CTP/dCTP (Supplementary information, Figure S9C), the O1 of the α-phosphate group faces TM5, which together with TM1 and TM4, forms a pair of hydrophobic grooves. These two grooves are clearly embedded within the membrane, which promotes binding of the hydrophobic tails of DGGGP (Figure 6D and 6E; Supplementary information, Figure S9C). This interaction is expected to position the phosphate head of the substrate adjacent to the α-phosphate group of CTP/dCTP (Figure 6A), enabling nucleophilic attack on the phosphate head (Figure 3A and 3B). This mechanism is supported by the observation that the mutation of critical residues around this region led to the loss of ApCarS activity (Figures 4 and 6). The catalytic mechanism of ApCarS is reminiscent of that of the enzyme UbiA, which includes three steps: acceptor ionization, condensation of isoprenoids, and product release22,23. In contrast to UbiA, ApCarS uses two ether-bonded isoprenoid chains at the condensation-like step.

Working model of the ApCarS catalytic reaction. (A) The proposed ApCarS CTP transferase reaction mechanism. Schematic depicting a putative three-step catalytic mechanism for DGGGP modification: (I) polarization, (II) condensation-like reaction, and (III) release. Residues critical to the process are highlighted. CTP and DGGGP are ApCarS substrates, and the large red arrow indicates nucleophilic attack. (B) Potential structural mechanism underlying ApCarS catalytic activity. Dual substrate (DGGGP and CTP) binding is associated with the repositioning of TM5, CL1 and CL2 at different states (O, open state; T, tight state). Modification of archaeol at the active site in the TM helix bundle drives the transition from O to T, causing CL1 and CL2 to move toward the CTP-binding pocket and the C-terminus of TM5 to move upwards in the DGGGP-binding pocket.
As mentioned above, TmCdsA and ApCarS share structural similarity with regard to their core catalytic domains (Figure 5A and Supplementary information, Figure S9A), and both utilize CTP as a substrate; however, they differ in the binding location of K+ and Mg2+ as well as in the recognition of the lipophilic substrate (Figure 5B and 5C; Supplementary information, Figure S9B). Our data showed that Mg2+ or Mn2+ is essential for ApCarS activity, whereas K+ and Li+ can enhance ApCarS activity in the presence of Mg2+ but not Mn2+. This latter ion specificity distinguishes ApCarS from other members of the CDS family (Supplementary information, Figure S4A), which use ions such as Ca2+ or Fe2+ instead of Mg2+21,24. The molecular basis for this difference, however, remains unclear.
The catalytic process of ApCarS appears to depend on conformational changes in CL1, CL2 and TM5 (Figure 7B and Supplementary information, Figure S9A). Given the higher flexibility of TM5 compared with the other ApCarS transmembrane helices (Supplementary information, Figure S10), as well as its involvement in the formation of the two potential archaeol-binding grooves, we hypothesize that TM5 functions by regulating lipophilic substrate entry or product release, as previously proposed21. This mechanism is consistent with the observation that the enzymatic activity of ApCarS is enhanced at elevated temperatures (Figure 1A), although other catalytic steps are potentially temperature dependent, such as enhanced diffusion rates of substrates and increased chances for substrate-enzyme collision. Efficient dissociation of pyrophosphate and release of CDP-archaeol from the active site are likely important for the optimal catalytic activity of ApCarS. In this respect, the structural flexibility of both TM5 and CPD (CL1 and CL2) may be important (Figure 7B and Supplementary information, Figure S5).
ApCarS and other CDP-archaeol synthases share only a low level of sequence identity with bacterial CDP-DAG synthase (Supplementary information, Figure S2), and these enzymes are highly specific for their respective substrates, DGGGP and phosphatidic acid20. Phylogenetic analysis demonstrated that both enzymes belong to the CTP transferase superfamily but are only distantly related, as they cluster in distinct subfamilies (Supplementary information, Figure S1). In contrast, the catalytic core domains of these proteins are structurally similar to each other. This finding supports an earlier proposal of the vertical inheritance of a CTP transferase gene from a cenancestor13. Possibly, this progenitor was less specific with respect to its lipophilic substrate (Figure 5 and Supplementary information, Figure S2). However, whereas the catalytic core domains of these enzymes remained largely conserved, the membrane-embedded substrate-binding domains evolved into two distinct subfamilies of CTP transferases.
In summary, we report the first crystal structure of the archaeal CarS family. Our data provide new insights into the general catalytic mechanism of transmembrane CTP transferases and the specific recognition of DGGGP by CarS. Additionally, our structural data provide new insights into the evolution of the distinct kingdoms of life, supporting the view that bacterial and archaeal transmembrane CTP transferases evolved from a common, likely less specific, ancestral enzyme.
Materials and Methods
Protein expression and purification
The codon-optimized gene encoding CarS from A. pernix K1 (ApCarS) was subcloned into the pET21b vector (Novagen, Madison, WI, USA). The 6×His-tagged protein was then expressed in E. coli BL21 (DE3) cells, and the cell membranes were harvested, solubilized in 1% dodecyl-β-D-maltopyranoside (DDM), and subsequently incubated with nickel-NTA resin at 4 °C for 1-2 h. The resin was washed in three steps with 15, 30 and 50 mM imidazole in buffer containing 15 mM Tris-HCl (pH 8.0), 350 mM NaCl, 10% glycerol and 0.03% DDM. Eluted proteins in DDM buffer were then further purified by size-exclusion chromatography, using a Superdex-200 column (GE Healthcare, Little Chalfont, UK), in 10 mM Tris-HCl (pH 8.0) buffer containing 0.02% DDM, 100 mM NaCl and 10% glycerol. In addition, to screen crystal formation in the presence of different detergents, ApCarS was subjected to size-exclusion chromatography in 10 mM Tris-HCl (pH 8.0) containing 100 mM NaCl, 10% glycerol, and either 0.05% n-dodecyl-N,N-dimethylamine-N-oxide (LDAO), 1% n-octyl-β-D-glucopyranoside (OG), 0.06% 6-cyclohexyl-1-hexyl-β-D-maltoside (CYMAL6), or 0.25% 5-cyclohexyl-1-pentyl-β-D-maltoside (CYMAL5). Subsequently, the purified fractions were pooled and flash-frozen for crystallization.
Crystallization
Each purified protein was concentrated to ∼25 mg/ml. Only the protein in complex with DDM could grow crystals in crystallization buffer. Crystals were grown using the LCP method25 at 20 °C in buffer containing 200 mM NaCl, 20% (w/v) PEG400, and 100 mM Tris-HCl (pH 8.0). The crystals (hexagonal plates) grew to a size of 0.03 × 0.1× 0.1 mm3 within 3 weeks.
Structure determination
X-ray diffraction data were collected at beamline BL19U1 of the Shanghai Synchrotron Radiation Facility. Data reduction was performed using the autoPROC pipeline26. The integration and scaling programs were XDS27 and AIMLESS28, respectively. The highest resolution limit was estimated using CC1/2 > 0.3, and I/sigma > 2. The initial phases were determined by ARCIMBOLDO_LITE29, a program that can determine phases using Phaser and SHELXE without homologous protein structures. Four α-helices of 25 residues were used as an initial estimate in the ARCIMBOLDO_LITE calculation. Then, a model was built using ARP/wARP30 software. Maximum likelihood-based refinement of the model was performed with Phenix31, and the atomic model was fitted using the Coot32 program. The stereochemical quality of the final model was assessed with MolProbity33.
In vitro activity assays and high-performance liquid chromatography-mass spectrometry analysis
The activity of wild-type and variant ApCarS proteins was examined in vitro as described previously20. For these assays, purified ApCarS (0.5 μM) was incubated in a reaction buffer containing 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 75 mM NaCl, 0.1% DDM, 125 mM imidazole and 5% glycerol, and enzymatic activity toward the chemically synthetic substrate DGGGP was evaluated in the presence of 2 mM CTP as another substrate. For wild-type ApCarS, reaction mixtures were incubated for 1 h at three different temperatures: 37 °C, 60 °C and 90 °C. In contrast, the activity of ApCarS variants was examined only at 90 °C. As specified, 20 mM EDTA was added to the reaction buffer to assess the importance of divalent ions. To test the ion requirement for ApCarS enzymatic activity, MgCl2, MnCl2, CaCl2 and ZnCl2 in combination with KCl, NaCl and LiCl were used as indicated in the figures (the concentration of each ion was 10 mM). Kinetic assays were performed by incubation in the same reaction buffer supplemented with 10 mM KCl, using increasing concentrations of CTP as indicated in the figure at 90 °C for 1 h. Reaction products were analyzed by high-performance liquid chromatography-mass spectrometry (HPLC-MS), as described in our previous work20.
ITC measurements
ITC measurements for the CTP-binding activity of wild-type and variant ApCarS proteins were taken at 25 °C using an ITC200 microcalorimeter (MicroCal (Malvern Instruments), Malvern, UK). Protein samples and substrates were prepared in buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM NaCl and 0.02% DDM. Approximately 0.5 mM CTP was then titrated into buffer containing 0.15 mM ApCarS; the first injection was 0.5 μl in volume and subsequent injections were 2 μl. The solution was stirred at a rate of 750 rpm. The data were fitted to a one site-binding model using MicroCal ORIGIN software. The representative thermodynamic parameters for the titration are shown in Supplementary information, Table S2. All ITC measurements were performed in technical triplicate.
Limited proteolysis analysis of ApCarS
The stability of the structure of ApCarS following substrate binding was analyzed by limited proteolysis. ApCarS protein (3.5 mg/ml) was mixed with 0.03 mg/ml trypsin (Roche, Basel, Switzerland) in buffer containing 15 mM Tris-HCl (pH 7.5), 100 mM NaCl, 0.02% DDM and 3 mM CTP in the presence or absence of 10 mM MgCl2. After 30 min of incubation on ice, proteins from the mixture were separated by SDS-PAGE and visualized by Coomassie blue staining.
MD simulations
DGGGP (ligand) was docked into the structure of ApCarS using the GOLD 5.0 program34 with default settings and a defined active site of 8.769 Å. During the pose refinement stage, the CHARMM36 force field35 was applied for the receptor and membrane lipid, as was CGenFF36 for the ligand and CTP. A steepest descent algorithm was performed to optimize the initial docking complex structure. Subsequently, the binding complex was modeled into a dimyristoyl phosphatidylcholine (DMPC) bilayer membrane using CHARMM-GUI37, with 119 DMPC molecules and 7 632 water molecules added automatically. With all non-solvent heavy atoms restrained, the system was minimized, and then heated in a 200-ps MD simulation in an NVT ensemble, followed by an 800-ps NPT equilibration, during which the restraints on the ligand were gradually released until no restraints were applied to the ligand. Finally, 100-ns MD production simulation was performed in the NPT ensemble. All simulations were run using GROMACS 5.1.438.
Lipid binding assay
Membrane lipid strips were utilized as previously described39. Briefly, 0.4 μl (0.005 mM) of DGGGP was spotted onto Hybond-C extra membranes (GE Healthcare Life Sciences), and the membrane strips were dried using a hair dryer. The dried membranes were then blocked by incubation with 5% (w/v) skim milk in TBST buffer (10 mM Tris-HCl (pH 7.5), 100 mM NaCl, 0.02% DDM and 0.01% Tween 20) three times for 10 min each. Then, each strip was incubated with 0.08 μg of each protein overnight at 4 °C. Subsequently, the membrane strips were incubated with anti-6×His mouse monoclonal antibodies (Cat# 230001; Zen BioScience, Research Triangle Park, NC, USA) overnight at 4 °C. The membrane strips were then washed with blocking buffer 5 times for 10 min each and then incubated with HRP-conjugated anti-mouse antibodies (Cat#511103; Zen BioScience) for 1 h. After washing the membrane strips with blocking buffer 6 times for 10 min each, the protein-lipid complexes were detected using enhanced chemiluminescence reagents.
Sequence alignment
Multiple sequence alignments were generated using ClustalW software and were edited with the ESPript 3.0 program40.
Accession code
The atomic coordinates and structure factors for the reported crystal structure have been deposited in the Protein Data Bank (PDB) under the accession code 5GUF.
Author Contributions
WC designed the research. SR and JW made the constructs. SR and QY purified the proteins and performed the ITC and lipid binding assays. SR and BS grew and optimized the crystals. CD collected the data. FY and XZ determined the structure. QS, SQ and XG created the molecular docking. QF and NH performed the MD simulations. AC examined the enzyme activity and performed the mass spectrometry. WC, JH and AJMD analyzed the data. WC wrote the manuscript with contributions from other authors.
Competing Financial Interests
The authors declare no competing financial interests.
References
Koga Y, Morii H . Biosynthesis of ether-type polar lipids in archaea and evolutionary considerations. Microbiol Mol Biol Rev 2007; 71:97–120.
Woese CR, Fox GE . Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci USA 1977; 74:5088–5090.
Jain S, Caforio A, Driessen AJ . Biosynthesis of archaeal membrane ether lipids. Front Microbiol 2014; 5:641.
Schneiter R, Toulmay A . The role of lipids in the biogenesis of integral membrane proteins. Appl Microbiol Biotechnol 2007; 73:1224–1232.
Dowhan W, Bogdanov M . Lipid-dependent membrane protein topogenesis. Annu Rev Biochem 2009; 78:515–540.
Bogdanov M, Xie J, Dowhan W . Lipid-protein interactions drive membrane protein topogenesis in accordance with the positive inside rule. J Biol Chem 2009; 284:9637–9641.
Di Paolo G, De Camilli P . Phosphoinositides in cell regulation and membrane dynamics. Nature 2006; 443:651–657.
Wymann MP, Schneiter R . Lipid signalling in disease. Nat Rev Mol Cell Biol 2008; 9:162–176.
Chaurio RA, Janko C, Munoz LE, Frey B, Herrmann M, Gaipl US . Phospholipids: key players in apoptosis and immune regulation. Molecules 2009; 14:4892–4914.
Koga Y, Morii H . Recent advances in structural research on ether lipids from archaea including comparative and physiological aspects. Biosci Biotechnol Biochem 2005; 69:2019–2034.
Koga Y . Thermal adaptation of the archaeal and bacterial lipid membranes. Archaea 2012; 2012: 789652.
Lombard J, Lopez-Garcia P, Moreira D . The early evolution of lipid membranes and the three domains of life. Nat Rev Microbiol 2012; 10:507–515.
Lombard J, Lopez-Garcia P, Moreira D . Phylogenomic investigation of phospholipid synthesis in archaea. Archaea 2012; 2012: 630910.
Icho T, Sparrow CP, Raetz CR . Molecular cloning and sequencing of the gene for CDP-diglyceride synthetase of Escherichia coli. J Biol Chem 1985; 260:12078–12083.
Sparrow CP, Raetz CR . Purification and properties of the membrane-bound CDP-diglyceride synthetase from Escherichia coli. J Biol Chem 1985; 260:12084–12091.
Langley KE, Kennedy EP . Partial purification and properties of CTP:phosphatidic acid cytidylyltransferase from membranes of Escherichia coli. J Bacteriol 1978; 136:85–95.
Kelley MJ, Carman GM . Purification and characterization of CDP-diacylglycerol synthase from Saccharomyces cerevisiae. J Biol Chem 1987; 262:14563–14570.
Shen H, Heacock PN, Clancey CJ, Dowhan W . The CDS1 gene encoding CDP-diacylglycerol synthase in Saccharomyces cerevisiae is essential for cell growth. J Biol Chem 1996; 271:789–795.
Wu L, Niemeyer B, Colley N, Socolich M, Zuker CS . Regulation of PLC-mediated signalling in vivo by CDP-diacylglycerol synthase. Nature 1995; 373:216–222.
Jain S, Caforio A, Fodran P, Lolkema JS, Minnaard AJ, Driessen AJ . Identification of CDP-archaeol synthase, a missing link of ether lipid biosynthesis in Archaea. Chem Biol 2014; 21:1392–1401.
Liu X, Yin Y, Wu J, Liu Z . Structure and mechanism of an intramembrane liponucleotide synthetase central for phospholipid biosynthesis. Nat Commun 2014; 5:4244.
Cheng W, Li W . Structural insights into ubiquinone biosynthesis in membranes. Science 2014; 343:878–881.
Huang H, Levin EJ, Liu S, Bai Y, Lockless SW, Zhou M . Structure of a membrane-embedded prenyltransferase homologous to UBIAD1. PLoS Biol 2014; 12:e1001911.
Nigou J, Besra GS . Cytidine diphosphate-diacylglycerol synthesis in Mycobacterium smegmatis. Biochem J 2002; 367:157–162.
Caffrey M . Crystallizing membrane proteins for structure determination: use of lipidic mesophases. Annu Rev Biophys 2009; 38:29–51.
Vonrhein C, Flensburg C, Keller P, et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr 2011; 67:293–302.
Kabsch W . XDS. Acta Crystallogr D Biol Crystallogr 2010; 66:125–132.
Evans PR, Murshudov GN . How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr 2013; 69:1204–1214.
Sammito M, Millan C, Frieske D, Rodriguez-Freire E, Borges RJ, Uson I . ARCIMBOLDO_LITE: single-workstation implementation and use. Acta Crystallogr D Biol Crystallogr 2015; 71:1921–1930.
Langer G, Cohen SX, Lamzin VS, Perrakis A . Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc 2008; 3:1171–1179.
Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010; 66:213–221.
Emsley P, Lohkamp B, Scott WG, Cowtan K . Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010; 66:486–501.
Chen VB, Arendall WB, 3rd, Headd JJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 2010; 66:12–21.
Jones G, Willett P, Glen RC . Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J Mol Biol 1995; 245:43–53.
Best RB, Zhu X, Shim J, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J Chem Theory Comput 2012; 8:3257–3273.
Vanommeslaeghe K, MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J Chem Inf Model 2012; 52:3144–3154.
Lee J, Cheng X, Swails JM, et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput 2016; 12:405–413.
Pronk S, Pall S, Schulz R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013; 29:845–854.
Dowler S, Kular G, Alessi DR . Protein lipid overlay assay. Sci STKE 2002; 2002:pl6.
Robert X, Gouet P . Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 2014; 42:W320–324.
Acknowledgements
We thank the staff of the BL19U1 beamline at the National Center for Protein Sciences Shanghai (NCPSS) at the Shanghai Synchrotron Radiation Facility for assistance with data collection. We thank J Chai for critical comments. We thank Peter Fodran and Adriaan Minnaard for the synthesis of DGGGP. This work was funded by the National Natural Science Foundation of China (NSFC; 31570842), the National Young Thousand Talents Program of China, and the Sichuan Province Thousand Talents Program to WC. AC was supported by a research program from the biobased ecologically balanced sustainable industrial chemistry (BE-BASIC). Computational support was provided by the special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase; U1501501).
Author information
Authors and Affiliations
Corresponding authors
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Phylogenetic tree distribution of members of the archaeal and bacterial CTP transferases families. (PDF 423 kb)
Supplementary information, Figure S2
Amino acid sequence alignment of CarS homologs in three life kingdoms. (PDF 1042 kb)
Supplementary information, Figure S3
Detection of products catalyzed by ApCarS using high performance liquid chromatography-mass spectrometry (HPLC-MS). (PDF 195 kb)
Supplementary information, Figure S4
Activity assays of ApCarS in the presence of different ions. (PDF 281 kb)
Supplementary information, Figure S5
Stabilization of cytoplasmic loop 1 (CL1) is essential for the CTP-binding and catalytic activity of ApCarS mutants. (PDF 396 kb)
Supplementary information, Figure S6
Electron density maps for the structure of ApCarS bound to CTP and metal ions. (PDF 564 kb)
Supplementary information, Figure S7
The stabilization of the periplasmic region is required for the enzymatic activity of ApCarS. (PDF 302 kb)
Supplementary information, Figure S8
Isothermal titration calorimetry (ITC) measurements of the CTP-binding activity of ApCarS encoding amino acid substitutions in the cytoplasmic domain (CPD). (PDF 848 kb)
Supplementary information, Figure S9
Structural basis for this catalytic mechanism. (PDF 900 kb)
Supplementary information, Figure S10
Structural analysis the potential binding site of Archaeol. (PDF 208 kb)
Supplementary information, Table S1
Data collect and refinement statistics (PDF 116 kb)
Supplementary information, Table S2
Representative affinity constants and binding parameters demonstrating the effect of amino acid substitutions within the cytoplasmic domain (CPD) of ApCarS on CTP-binding (PDF 70 kb)
Rights and permissions
About this article

Cite this article
Ren, S., Caforio, A., Yang, Q. et al. Structural and mechanistic insights into the biosynthesis of CDP-archaeol in membranes. Cell Res 27, 1378–1391 (2017). https://doi.org/10.1038/cr.2017.122
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2017.122
Keywords
This article is cited by
-
The catalytic and structural basis of archaeal glycerophospholipid biosynthesis
Extremophiles (2022)
